Nucleosides and nucleoside analogues, pharmaceutical composition and processes for the preparation of the compounds
Field of the invention
The present invention relates to the use of chemical compounds and physiologically acceptable salts thereof for the therapheutic and prophylactic control and treatment of the Acquired Immuno Deficiency
Syndrome (AIDS), infections by Human Immunodeficiency Virus, hepatitis B virus infections and retrovirus infections and method for such control and treatment in animal and man.
Background of the invention
In the late seventies a new disease was reported, which subsequently was referred to as Acquired Immuno Deficiency Syndrome (AIDS). It is now generally accepted that a retrovirus referred to as HIV (Human Immunodeficiency Virus), formerly known as Human T-cell Lymphotropic Virus
(HTLV-III) or Lymphadenopathy Associated Virus (LAV) plays an essential role in the etiology of AIDS.
AIDS is characterized by a profound immunodeficiency due to low numbers of a subset of lymphocyte-T-helper cells, which are one target for HIV infection. The profound immunodeficiency in AIDS patients makes these, patients highly susceptible to a variety of opportunistic infections of bacterial, fungal, protozoa! or viral etiology. The etiological agents among viral opportunistic infections are often found in the herpes virus group, i.e., Herpes simplex virus (HSV), Varicella Zoster virus (VZV), Epstein-Barr virus (EBV) and, especially, cytomegalovirus (CMV). Other retroviruses affecting humans are HTLV-I and II and examples of retroviruses affecting animals are feline leukemia virus and equine infectios anaemia virus.
Hepatitis B virus infections cause severe disease such as acute hepatitis, chronic hepatitis, fulminant hepatitis in a considerable number of persons. It is estimated that there are 200 million patients with chronic hepatitis B infection in the world. A considerable number of the
chronic cases progress to liver cirrosis and liver tumours. In some cases the hepatitis infections also take a rapid and severe course as in fulminant B hepatitis with about 90 % mortality. At present there is no known effective treatment against hepatitis B infections.
General outline of the invention
A great number of nucleoside analogues exhibit several antimetabolic activities. They do so by substituting for or competing with the naturally occuring nucleosides. Recently some nucleoside analogues have been described, which inhibit in cell culture the multiplication of human immunodeficiency virus (HIV, also called HTLV-III, LAV), the causative agent of AIDS and AIDS-related complex (ARC). Such compounds are for example azidothymidine, dideoxycytidine and dideoxyadenosine. These and other described HIV-antimetabolic nucleoside analogues have the same geometric relationship between the nucleoside base and the glycosidic part as the naturally occuring nucleosides, i.e. they are β-anomers.
We have now, surprisingly, found that some nucleosides and nucleoside analogues with the opposite geometric configuration, α-anomers, are potent inhibitors of HIV multiplication but not of cell-division. Anti-HIV activities are displayed by such geometric isomers which have been modified either in the nucleoside base part, the glycoside part or in both parts. The structures of these compounds are disclosed in this invention.
Prior Art
The following compounds of the formula I below are known:
1. Compounds of the formula
wherein R
3 is OH and R
1 is as follows:
R1 is H and CH3: T. Nishimura, B. Shinizu, I. Iwai
Chem. Pharm. Bull. (Tokyo) 12 (1964), 1471
is C2H5: M. Swierkowski, D. Shugar J. Med. Chem. 12 (1969), 533
R1 is n-C3H7: A. Szaboles, J. Sagi, L. Otvös
J. Carbohydrates, Nucleosides, Nucleotides 2 (1975), 197 - 211
R1 is i-C3H7: M. Draminski, A. Zgit-Wroblewska Polish J. Chemistry 54 (1980), 1085
R1 is C≡CH: P.J. Barr, A.S. Jones, P. Serafinowski, R. Walker J. Chem. Soc. Perkin I (1978), 1263 - 1267
and wherein R3 is N3 and R1 is CH3: M. Imezawa, F. Eckstein, J. Org, Chem. 43 (1978), 3044-3048.
2. The compound of the formula
R1 is C≡CH is described by P.J. Barr, A.S. Jones, P. Serafinowski, R. Walker, J. Chem. Soc. Perkin I (1978), 1253 - 1267
R1 is H is described by J.J. Fox, N.C. Yung, I. Wempen and M. Hoffer, J. Am. Chem. Soc, Vol. 83 (1961), 4066-4070.
Both groups 1. and 2. concern only compounds having the 3' group and the 4'hydroymethyl group in a trans-configuration.
Disclosure of the invention
It has been found according to the present invention that the compounds of the formula
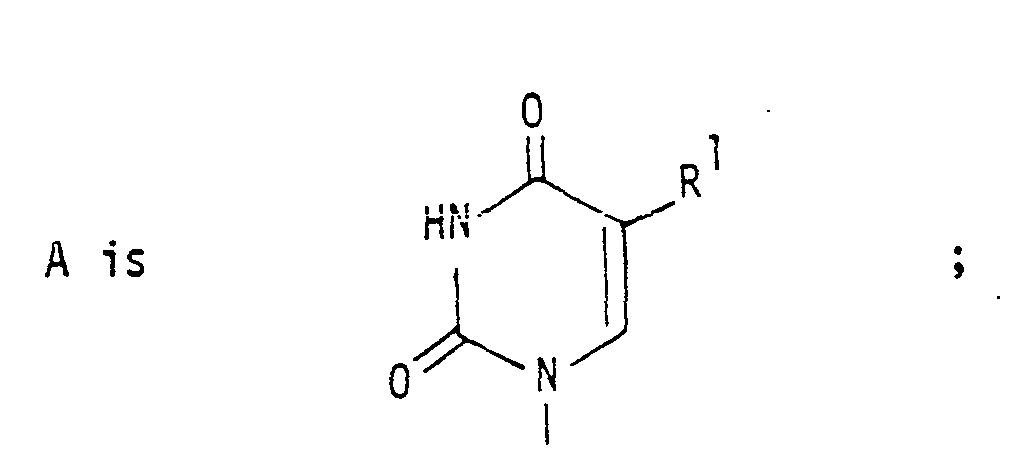
wherein the radicals A, X, R1, R2 and R2 are defined as follows:
X: (a) O
(b) S
(c) CH2
R1 : H ; alkyl containing 1-3 carbon atoms ;
-CH=CH2 ;-CH=CH-CH3;-CH2-CH=CH2 ; -C-CH3 ;-C≡ CH
R2: H; or R constitutes together with R3 a carbon-carbon bond
R3: H; F; Cl; Br; I; N3; CN;-C≡CH; OH; OCH3; CH2OH; or R3 constitutes together with R2 a carbon-carbon bond,
and therapeutically acceptable salts thereof, inhibit the multiplication of human immunodeficiency virus (HIV). The compounds of the formula I are useful as therapeutic and/or prophylactic agents in the control and treatment of HIV virus infections in mammals and man.
In a more general aspect, the compounds of the formula I are useful as therapeutic and/or prophylactic agents in the control and treatment of infections caused by retroviruses and hepatitis B virus in mammals and man.
All retroviruses, including HIV, require the enzyme reverse transcriptase in their natural cycle of replication.
Hepatitis B virus (HBV) is a DNA virus with a unique circular doublestranded DNA genome which is partly single-stranded. It contains a specific DNA polymerase required for viral replication. This DNA polymerase also acts as a reverse transcriptase during the replication of HBV DNA via an RNA intermediate.
The compounds of the formula I inhibit the activity of reverse transcriptase of retroviruses including HIV as well as the activity of DNA polymerase of hepatitis B virus.
The present invention has several aspects:
1. the novel compounds included in the formula I,
2. pharmaceutical compositions comprising a compound of the formula I as active ingredient,
3. a compound of the formula I for use in therapy,
a compound of the formula I for use in the manufacture of a medicament for therapeutic and/or prophylactic treatment of infections caused by a retrovirus, including HIV, or by hepatitis B virus,
a method for the therapeutic and/or prophylactic treatment of infections in mammals and man caused by retrovirus including HIV or hepatitis B virus, by administering to a host in need of such treatment an efficient amount of a compound of the formula I.
It is a preferred aspect of the invention to combat HIV virus infections in man.
The expression "alkyl containing 1-3 carbon atoms" for the radical R1 means CH3, C2H5, CH2CH2CH3, CH(CH3)2 and cyclopropyl.
When R3 in formula I is F, Cl, Br, I, N3, CN, C≡CH, OH, OCH3 or CH2OH it may have either cis-configuration or trans-configuration relative to the hydroxymethyl function at position 4'.
Preferred compounds of the formula I are:
(c) R
3 at position 3' and the hydroxymethyl group at position 4' have the trans-configuration
(d) R1 is CH3 or C2H5
(e) X is O or CH2
(f) X is O
(g) R2 is H
(h) R2 constitutes together with R3 a carbon-carbon bond
(i) R3 is H, F, N3, OH, OCH3, or CH2OH or constitutes together with R2 a carbon-carbon bond
(j) R3 is H, F, or N3
(k) the combination of (a), (c), (d) and (e) above
(l) the combination of (a), (c), (d), (e), (g) and (i) above
(m) the combination of (a), (c), (d), (f), (g) and (j) above
(n) the combination (a), (c), (d), (e) and (h) above
(o) the combination (b), (c), (d) and (e) above
(p) the combination (b), (c), (d), (e), (g) and (i) above
(q) the combination (b), (c), (d), (f), (g) and (j) above
(r) the combination (b), (c), (d), (e) and (h) above
Examples of preferred compounds are:
R
1 is CH
3 R
2 is H; R
3 is H R
1 is CH
3 R
2 is H; R
3 is OH R
1 is CH
3 R
2 is H; R
3 is OCH
3 R
1 is CH
3 R
2 is H; R
3 is CH
2OH R
1 is C
H3 R
2 is H; R
3 is F R
1 is CH
3 R
2 is H; R
3 is N
3 R
1 is CH
3 R
2 and R
3 constitute together a carbon-carbon bond
R1 is C2H5; R2 is H; R3 is H R1 is C2H5 R2 is H; R3 is OH R1 is C2H5 R2 is H; R3 is OCH3 R1 is C2H5 R2 is H; R3 is CH2OH R1 is C2H5; R2 is H; R3 is F R1 is C2H5 R2 is H; R3 is N3 R1 is C2H5 R2 and R3 constitute together a carbon-carbon bond
R2 is H; R3 is OH
R2 is H; R3 is OCH3
R2 is H; R3 is CH2OH R2 is H; R3 is F
R2 is H; R3 is N,
R2 and R3 constitute together a chemical bond
In all the examples of preferred compounds R3 at position 3' and hydroxymethyl at position 4' have the trans-configuration.
In clinical practice the nucleosides of the formula I will normally be administered orally, by injection or by infusion in the form of a pharmaceutical preparation comprising the active ingredient in the form of the original compound or optionally in the form of a pharmaceuticall acceptable salt thereof, in association with a pharmaceutically acceptable carrier which may be a solid, semi-solid or liquid diluent or an ingestible capsule. The compound may also be used without carrier material. As examples of pharmaceutical preparations may be mentioned tablets, dragέes, capsules, granulates, suspensions, elixirs, syrups, solutions etc. Usually the active substance will comprise between 0.05 and 20 % for preparations intended for injection and between 10 and 90 % for preparations intended for oral administration.
In the treatment of patients suffering from retrovirus, especially HIV, or hepatitis B virus infections, it will be preferred to administer the compounds by any suitable route including the oral, parenteral, rectal, nasal, topical and vaginal route. The parenteral route includes subcutaneous, intramuscular, intravenous and sublingual administration. The topical route includes buccal and sublingual administration. The dosage at which the active ingredients are administered may vary within a wide range and will depend on various factors such as the severity of the infection, the age of patient etc., and may have to be individually adjusted. As a possible range for the amount of the compounds of the invention or a physiologically acceptable salt thereof to be administered per day may be mentioned from about 10 mg to about 10 000 mg. pre
ferentially 100 - 500 mg for intravenous administration and preferentially 100 - 3000 mg for oral administration.
Examples of pharmaceutically acceptable salts of the compounds of formula I include base salts, e.g. derived from an appropriate base, such as alkali me l (e.g. sodium), alkaline earth metal (e.g. magnesium) salts, ammonium and NX4 + (wherein X is C1-4 alkyl). Physiologically acceptable salts of a hydrogen atom or an amino group include salts of organic carboxylic acids such as acetic, lactic, gluconic, citric, tartaric, maleic, malic, panthothenic, isethionic, succinic, oxalic, lactobionic and succinic acids; organic sulfonic acids such as methanesulfonic, ethanesulfonic, benzenesulfonic, p-chlorobenzenesulphonic and p-toluenesulfonic acids and inorganic acids such as hydrochloric, hydroiodic, sulfuric, phosphoric and sulfamic acids. Physiologically acceptable salts of a compound of an hydroxy group include the anion of said compound in combination with a suitable cation such as Na+, NH4 +, and HX4 +
(wherein X is a C1-4 alkyl group).
Those compounds of the formula I which are novel are summarized as compounds of the formula I with the provisos that
1. when A, X, R2 and R3 are combined as follows:
X is O; R2 is H; R3 is OH;
then R
1 is -CH=CH
2, -CH=CH-CH
3, -CH
2-CH-CH
2, or cyclopropyl;
2. when A, X, R
2 and R
3 are combined as follows:
X i s O ; R2 i s H ; R3 i s N3 ;
then R
1 is H; alkyl containing 2-3 carbon atoms, -CH=CH
2; -CH=CH-CH
3; -CH
2-CH=CH
2; -C≡CH; or cyclopropyl;
3. when A, X, R
2 and R
3 are combined as follows:
X is O; R
2 is H; R
3 is OH;
then R
1 is alkyl containing 1-3 carbon atoms, -CH=CH
2; -CH=CH-CH
3; -CH
2-CH=CH
2; or cyclopropyl.
The administered compounds may also be used in therapy in conjunction with other medicaments such as 9-[(2-hydroxy-1-(hydroxymethyl)ethoxy)- methyl]guanine, 9-(2-hydroxyethoxymethyl)guanine (acyclovir), 2-amino- 9-(2-hydroxyethoxymethyl)purine, interferon, e.g., α-interferon, interleukin II, and phosphonoformate, or in conjunction with immune modu
lating therapy including bone marrow or lymphocyte transplants or medications such as levamisol or thymosin which would increase lymphocyte numbers and/or function as is appropriate.
Methods of preparation
The compounds of the invention may be prepared by one of the following general methods, constituting a further aspect of the invention.
A. Condensing a glycoside as comprised in formula I, where the hydroxyl groups may be optionally protected, to the N-1 position of a pyrimidine derivative, corresponding to radical A in formula I according to known methods described in the literature, followed by separation of the α-anomer and removal of any protecting group (s). Such methods are described for example in "Basic Principles in Nucleic Acid Chemistry", Vol. 1 (Academic Press, 1974, Ed. P.O.P.Ts'o), in "Nucleoside Analogues, Chemistry, Biology and Medical Applications" (Pharma Press, 1979, Eds. R.T. Walker, E. De Clercq and F. Eckstein) and in Nucleic Acids Research Vol. 12, 1984, pages 6827 - 6837 (A.J. Hubbard, A.S. Jones and R.T. Walker). An example of such a method is given for
the case of a uracil base analogue:
wherein R4 is H, F, Cl, Br, I, N3, CN, C≡CH, OR5, OCH3 or CH2OR5, R5 is a protecting group, of which a great variety is known, and examples of which are p-toluoyl, acetyl, trityl, benzyl. R1 and R2 are as defined above.
B . Anomerization of a β-anomer of the formula
wherein A, X, and R
2 are as defined above, R
4 is H; F; Cl; Br; I; N
3;
CN; OR5; OCH3; or CH2OR5; wherein R5 is H or a hydroxy-protecting group to a mixture of α- and β-anomers, whereafter the α-anorner is separated and any protecting groups removed. The anomerization may be performed by known methods, e.g. with an optionally protected β-nucleoside, for example a silylated nucleoside, with a catalyst, such as for example trimethylsilyl trifluoromethanesulfonate
R1, R2, R4 and R5 are as defined above,
C. A transglycosylation reaction whereby the sugar moiety forming a bond, α- or β-, to one nucleoside base, is transferred to the desired pyrimidine base. The reaction is performed with a catalyst such as for example trimethylsilyl trifluoromethanesulfonate, and is followed by separation of the products and deprotection.
wherein R1, R2, R4 and R5 are as defined above. The radical B is a pyrimidine or purin base, the choice of which is not critical.
D. Introduction of the functional group R3, or a precursor of R3, into the nucleoside α-anomer by substitution of a suitable leaving group, R7 , followed by deprotection.
R
1 and R
5 are as defined above, R
7 is a good leaving group such as for example trifluoromethanesulfonyloxy, R
8 is F, Cl, Br, I, N
3, CN,
OCH3 and synthos for the C≡CH, OH and CH2OH groups, such as for example
C≡C-Si(CH
3)
3, CH
3CO
2 and R
9 is a suitable protecting group,
An alternative way for introduction of the R8 function is by reaction of the 2,3'-anhydroα-anomer.
wherein R
1, R
5 and R
8 are as defined above.
The principles of methods A-D above are applicable to the synthesis of both uridine and cytidine analogues of formulas I and II, although the formulas illustrating the reactions only depict uridine analogues.
E. Converting the uracil moiety of the 5-substituted or unsubstituted α-uridine compounds to a cytosine moiety of the corresponding α-cytidine analogues. This is carried out by conventional methods, the principles of which have been described for example by W.L. Sung (J. Chem. Soc. Chem. Commun. 1981, p. 1089 and J. Organic Chemistry 1982, volume 47, pages 3623 - 3628) and by P. Herdewijn et al. (J. Medicinal Chemistry 1985, volume 28, pages 550 - 555).
The following examples will further illustrate the invention.
Preparation of intermediate products
A. Preparation of ST-3'-deoxy-5'-0-acetylthymidine (VSB423)
3'F-3'-deoxythymidine 45 mg (0.184 mmol) in acetic anhydride (2.0 mL) was heated with stirring in an oil bath at 80° for 7 hrs. The solution was evaporated in vacuo and the residual acetic anhydride and acetic acid were removed by several additions and reevaporations with benzenetoluene (1:1). The residue was used without further purifications.
Preparation of compounds of the invention
Example 1. Preparation of 1-(3-F-2,3-dideoxy-α-D-ribofuranosyl)thymine (VSA 419) (Method B)
Thymine 23 mg (0.18 mmol) and 3'F-3 -deoxy-5'-0'-acetylthymidine was suspended in acetonitrile (1.2 mL) and N,0-Bis (trimethylsilyl)-acetamide (0.35 mL) was added. The mixture was stirred at room temperature for 1.5 hrs. Trimethylsilyl trifluoromethanesulfonate (0.05 mL) was added. After stirring at room temperature for 192 hrs, the mixture was poured under stirring into a 1:1 (v/v) mixture of 20 ml of 10 % aqueous KHCO3-ethyl acetate. Two phases were separated and the water phase was extracted with ethyl acetate (3 x 10 mL). The combined ethyl acetate phase was filtered and evaporated in vacuo. The residue was dissolved in dichloromethane-ethyl acetate 1:1 and applied to a column of silica gel, and the column was eluated with dichloromethane-ethyl acetate 1:1 to give 28 mg (53 %) starting material (VSB 423) (Rf 0.37 on TLC silicagel CH2Cl2-EtoAc 1:1) and 18 mg (34 %) of 1-(3-F-2,3-dideoxy-5-0-acetyI-α-D-ribofuranosyl)thymine (VSB 424) (Rf 0.29 on TLC silica gel CH2Cl2-EtoAc 1:1).
NMR (CD3OD) tfl.95 (s, 3H, CH3-5), 2.12 (s, 3H, CH3CO), 2.3-3.0 (m, 2H, H-2'a,b), 4.15 (d, 2H, J4',5'=4.4 Hz, H-5'a,b), 4.81 (dt, 1H, J3'F,4'=30.0 Hz, J4',5'=4.6 Hz, H-4'), 5.23 (dd, 1H, J3'F,3'=53.7 Hz, J2',3'=5.0, H-3'), 6.36 (d, 1H, J1',2'=7.57 Hz, H-1'), 7.27 (d, 1H, J H-6, CH3=1.47, H-6)
13C(CD3OD) δ12.80 (CH3), 20.87 (CH3CO), 39.40 (d, J=20.8 Hz, C-2'), 63.37 (d, J=12.2 Hz, C-5'), 84.65 (d, J=24.4 Hz, C-4'), 86.50 (s, C-1') 93.82 (d, J=178 Hz, C-3'), 111.12 (C-5), 135.07 (d, J=6.1 Hz, C-6), 150.48 (C-2), 163.68 (C-4), 170.30 (CH3CO).
The compound VSB 424 (16 mg) was dissolved in saturated methanolic ammonia (5 mL) and left at room temperature overnight. The solution was evaporated and the residue was treated with acetone-benzene (1:4) to give crystals of the desired compound, VSA 419 (9.4 mg, 69 %) UV λ max (H2O) 269 nm.
NMR (DMSO-d6) Η δ1.79 (d, 3H, J CH3, H-6=1.2 Hz CH3), 2.16-2.90 (m, 2H, H-2'), 3.2-3.6 (m, 2H, H-5'), 4,61 (dt, 1H, J3'F,4'=23.4 Hz, J4',5' ~4 Hz, H-4'), 5.06 (t, 1H, J5',OH=5.6 Hz, OH), 5.32 (dd, 1H, J3'F,3'=54.2 Hz J2'3'=4.9 Hz, H-3'), 6.18 (dd, 1H, J1'2'=7.7 Hz and 2.1 Hz, H-T), 7.39 (d, 1H, J CH3, H-6=1.2 Hz, H-6) 13C (DMSO-d6) (δ12.46 (CH3),~39 (C-2'), 61.17 (d, J=11.0 Hz, C-5'), 85.85 (C-1'), 87.15 (d, J=20.8 Hz, C-4'), 94.75 (d, J=173 Hz, C-3'), 109.15 (C-5), 135.63 (d, J=6.1 Hz, C-6), 150.53 (C-2), 163.95 (C-4)
Example 2. Preparation of 1-(3-F-2,3-dideoxy-α-D-ribofuranosyl)-5-propyluracil (VSA 409) (Method C)
5-Propyluracil (56 mg) and 3'-F-3'deoxythymidine (47 mg) were suspended in acetonitrile (1.2 mL) and N,O-Bis (trimethylsilyl) acetamide
(0.35 mL) was added. The mixture was stirred at room temperature for 1.5 hrs. Trimethylsilyl trifluoromethanesulfonate (0.05 mL) was added. After stirring at room temperature for 138 hrs, the mixture was evaporated in vacuo and added to H2O (0.5 mL), filtered and washed with H2O (0.5 nl). The combined water phase was applied to a C18-column (HPLC) and eluted with methanol-water (35:65), at a rate of 7.0 ml/min. The β -anomer eluted after 12.9 min, and the desired α-anomer, VSA 409, after 18.0 min. Yield 9.3 mg (18 %), UV λ max (H2O) 269 nm, MS M+272 (10 %) , 154 (100 %), 119 (76 %) .
Example 3. Preparation of 1-(3-F-2,3-dideoxy-α-D-ribofuranosyl)-5-ethyluracil (VSA 411) (Method C)
5-Ethyluracil (51 mg) and 3'F-3'-deoxythymidine (48 mg) were suspended in a tonitrile (1.2 mL) and N,O-Bis (trimethylsilyl) acetamide
(0.35 mL) was added. The mixture was stirred at room temperature for 1.5 hrs. Trimethylsilyl trifluoromethanesulfonate (0.05 mL) was added. After stirring at room temperature for 161 hrs, the mixture was evaporated in vacuo, and added to water (0.5 mL), filtered and washed with water (0.5 mL). The combined water phase was applied to a C18-column
(HPLC) and eluted with methanol-water (1:3) at a rate of 8.0 ml/min. The β-anomer eluted after 12.3 min and the desired α-anomer, VSA 411, after 16.4 min. Yield 13.1 mg (26 %) . UV λmax (H2O) 267.5 nm. MS M+ 258
(9 %) , 140 (100 %), 119 (67 %) .
Example 4. Preparation of 1-(2-deoxy-α-D-ribofuranosyl)-5-isopropenyluracil (VSA 175) (Method A)
5-Isopropenyluracil (4.3 g), hexamethyldisilazane (50 ml), chlorotrimethylsilane (1 ml) and ammoniumsulfate (catalytic amount) were heated at reflux for 2.5 hrs. Excess of solvent was evaporated in vacuo and the residual bis-silylated 5-isopropenyluracil (8.4 g.) was dissolved in dichloroethane (50 ml) and added to 2-deoxy-3,5-di-0-p-toluoyl-D-erythro-pentosyl chloride (11.0 g) in dichloroethane (150 nil) also containing molecular sieves (4 A, 15 g). The suspension was stirred at room temperature overnight, after which it was filtered and the solvent was evaporated. The residue was redissolved in dichloromethane which was washed with saturated aq NaHCO3 and H2O, dried over Na2SO4 and concentrated to a volume of about 70 ml. A precipitate formed which was filtered off, dichloromethane was evaporated from the filtrate and the residue was subjected to chromatography on silica gel columns eluted with hexane/ethylacetate/dichloromethane (5/5/3), to give 1-(2-deoxy3,5-di-0-p-toluoyl-α-D-ribofuranosyl)-5-isopropenyluracil (VSA 174), 2.64 g (Thin layer chromatography, silica gel, solvent system as above, Rf=0.5).
Sodium metal (0.25 g) was dissolved in dry methanol (263 ml), compound VSA 174 (2.64 g) was added and the solution was stirred at room temperature overnight, after which water (35 ml) was added. The solution was neutralized with an ion exchanger (Dowex H 50Wx2), filtered and the solvent was evaporated. The residue was washed with hexane and purified by chromatography on a column of silica RP18 eluted with 50 % aq methanol to give 1-(2-deoxy-α-D-ribofuranosyl)-5-isopropenyluracil. (TLC silica RP8, 50 % aq methanol, Rf=0.5).
Biological tests
Test I. Effect of compounds of the formula I on HIV in H9 cells
Materials and methods: HIV infection of H9 cells
H9 cells, 105 cells per well on a 24 well plate, suspended in 2 ml RPMI-medium containing 10 % fetal calf serum, 100 μg/ml penicillin, 10 μg/ml streptomycin sulfate and 2 μg/ml polybrene are exposed to HIV (HTLV-IIIn) and different concentrations of the test compounds. The plates are incubated at 37°C in 5 % CO2 for 6 - 7 days. The contents in each well is then homogenized with a pipette and transferred to a centrifuge tube. After centrifugation for 10 min at 1500 rpm the supernatent is removed and the cell pellet is analyzed by fixing in methanol on glass plates. Human HIV positive serum diluted 1:80 or 1:160 is adde and incubated for 30 min at 37°. The plate is then washed with phosphate-buffered saline (PBS) containing Ca2+ and Mg2+. Sheep antihuman conjugate (FITC) is added and after a new incubation the plate is again washed with PBS. Contrast staining is done with Evans blue and after drying the frequency of HIV antigen containing cells is determined in a microscope. The test result is shown in Table 1.
Table 1. Concentration (μM) for 50 % inhibition (IC50) of human immuno deficiency virus multiplication in cell culture
Compounds IC50(μM)
1-(3-fluoro-2,3-dideoxy-α-D-ribofuranosyl)-5-ethyluracil (VSA 411) 0.1
1-(3-fluoro-2,3-dideoxy-α-D-ribofuranosyl)-5-propyluracil (VSA 409) 2.5
1-(2-deoxy-α-D-ribofuranosyl)-5-ethyluracil (VIP 289) 10
It is seen in Table 1 that the tested compounds are active inhibitors of HIV virus multiplication.
Test II. Cellular toxicity
H9 cells, 2x107 cells per plate, are incubated in RPMI-1640 medium containing 10 % fetal calf serum, 70 mg/1 penicillin, 100 mg/1 streptomycin and 10 mM hepes, in absence or precence of test compounds. The number of cells per plate is determined after 48 hrs. Cells incubated in absence of test compound then underwent two cell division cycles.
F5000 cells, which are human embryo cells, 1x105 cells per plate, are incubated in Eagle's minimal essential medium, supplemented with Earle's salts, non-essential amino acids, 10 % fetal calf serum, 10 mM hepes, 70 mg/1 penicillin and 100 mg/1 streptomycin, in absence or presence of test compounds. The number of cells per plate is determined after 48 hrs. Cells incubated in absence of test compounds underwent one cell division cycle. The results are given as TC50, which is the concentration of a compound which gives 50 % inhibition of cell multiplication.
Table 2. Cellular toxicity and H9 and F5000 cells
-TC50(μM)
Compound H9 F5000
1-(3-fluoro-2,3-dideoxy-α-D-ribofuranosyl)-5-ethyluracil (VSA 411) 400 500
1-(3-fluoro-2,3-dideoxy-α-D-ribofuranosyl)-5-methyluracil (VSA 419) 250
1-(2-deoxy-α-D-ribofuranosyl)-5-ethyluracil (VIP 289) 1000
It is seen in Table 2 that the test compounds exhibit TC50 values which vastly exceed the concentration IC50 according to Table 1 to 50 % inhibition of HIV virus multiplication.