WO1996040705A1 - C-4' modified adenosine kinase inhibitors - Google Patents
C-4' modified adenosine kinase inhibitors Download PDFInfo
- Publication number
- WO1996040705A1 WO1996040705A1 PCT/US1996/010404 US9610404W WO9640705A1 WO 1996040705 A1 WO1996040705 A1 WO 1996040705A1 US 9610404 W US9610404 W US 9610404W WO 9640705 A1 WO9640705 A1 WO 9640705A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- phenyl
- amino
- halogen
- guanidino
- Prior art date
Links
- 0 *C([C@@]([C@@](C(N)=O)O*)O)N Chemical compound *C([C@@]([C@@](C(N)=O)O*)O)N 0.000 description 4
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/14—Pyrrolo-pyrimidine radicals
Definitions
- This invention relates to adenosine kinase inhibitors and to nucleoside analogs, C-4' modified pyrrolo[2,3-d] pyrimidine and pyrazolo[3,4-d] pyrimidine nucleoside analogs having activity as adenosine kinase inhibitors.
- the invention relates to nucleoside analogs of this kind, having zero substitutions or two substitutions at the C-4' position of the furanose (sugar) moiety.
- the invention also relates to the preparation and use of these adenosine kinase inhibitors in the treatment of cardiovascular, and cerebrovascular diseases, inflammation and other diseases which can be regulated by increasing the local concentration of adenosine.
- Adenosine is an endogenously produced molecule that plays a major role in a variety of important cellular processes. It is a vasodilator, can inhibit immune function, enhance activation of mast cells (associated with allergic reactions), inhibit neutrophil oxygen free-radical production, is antiarrhythmic, and is an inhibitory neurotransmitter. Adenosine is phosphorylated to adenosine triphosphate (ATP) which is used by all cells to store energy for use in future energy- utilizing metabolic reactions or mechanical work (e.g. muscle contraction).
- ATP adenosine triphosphate
- Extracellular adenosine frequently prouced by breakdown of intracellular ATP pools, evokes a variety of pharmacological responses through activation of extracellular adenosine receptors located on the surface of nearly all cells.
- adenosine produces a variety of cardiovascular related effects including vasodilation, inhibition of platelet aggregation, and negative inotropic, chronotropic and domotropic effects on the heart.
- Adenosine also has effects within the central nervous system (CNS) including inhibition of neurotransmitter release from presynaptic neurons and inhibition of post-synaptic neuron firing in brain and the spinal cord and at sites of inflammation, such as inhibition of neutrophil adhesion to endothelial cells and inhibition of neutrophil oxygen free-radical production.
- CNS central nervous system
- Compounds that increase extracellular adenosine can be beneficial to living organisms, particularly under certain conditions.
- compounds that increase adenosine levels have been associated with the treatment of ischemic conditions such as stroke, as well as other conditions benefitted by enhanced adenosine levels, such as inflammation, arthritis, seizures, epilepsy and other neurological conditions.
- the compounds are also useful for treating pain, as muscle relaxants, and for inducing sleep.
- Adenosine kinase is a cytosolic enzyme which catalyzes the phosphorylation of adenosine to AMP. Inhibition of adenosine kinase can potentially reduce the ability of the cell to utilize adenosine, leading to increased adenosine outside of the cell where it is pharmacologically active.
- the regulation of adenosine concentration is complex and involves other adenosine-metabolizing enzymes each with different kinetic properties and mechanisms of regulation.
- Adenosine can also be deaminated to inosine by adenosine deaminase (ADA) and condensed with L-homocysteine to S-adenosylhomocysteine (SAH) by SAH hydrolase.
- ADA adenosine deaminase
- SAH S-adenosylhomocysteine
- the role of each of these enzymes in modulating adenosine concentration is dependent on the prevailing physiological conditions, is tissue specific and is not well understood.
- a number of nucleosides including pyrrolo[2,3-d]pyrimidine and pyrazolo[3,4-d]pyrimidine analogs have been evaluated for inhibition of adenosine kinase but were reported to have K j 's of greater than 800 nM. Caldwell and Henderson. Cancer Chemother.
- adenosine release has been measured in neuroblastoma cells in culture and compared with that of a variant deficient in adenosine kinase (AK " ).
- the AK cells used in this study were said to release adenosine at an accelerated rate; the concentration of adenosine in the growth medium was reported to be elevated compared to the normal cells. Green, 1 Supramol. Structure. 13:175-182 (1980).
- adenosine uptake was reportedly inhibited by the adenosine kinase inhibitors, 5- iodotubercidin and 5'-deoxy-5-iodotubercidin. Davis et al., Biochem. Pharmacol.. 33:347-55 (1984). However, inhibition of uptake and intracellular trapping via phosphorylation does not necessarily result in increased extracellular adenosine, since the adenosine could enter other metabolic pathways or the percentage of adenosine being phosphorylated could be insignificant compared to the total adenosine removed.
- adenosine kinase inhibitors with a useful half-life, i.e. compounds which can be exploited to beneficially influence or control endogenous adenosine kinase activity, and therefore, extracellular adenosine levels.
- the compounds of the invention are suitable adenosine kinase inhibitors having these characteristics.
- the invention is directed to novel pyrrolo[2,3-d] pyrimidine or pyrazolo[3,4-d] pyrimidine nucleoside analogs having activity as adenosine kinase inhibitors, wherein the furanose moiety has zero substituents or two substituents at the C-4' position.
- Preferred substitutents are hydroxymethyl, aminomethyl, and methyl. Most preferred are compounds where both substituents are the same, but are not both methyl, or both substituents form a small ring, such as cyclopropyl.
- additional asymmetric carbons may be present in compounds of the present invention, for example in the substituted heterocyclic pyrrolo[2,3-d] pyrimidine or pyrazolo[3,4-d]pyrimidine ring. All of the resulting isomers, enantiomers, and diastereomers are considered to fall within the scope of the present invention.
- These compounds are selective adenosine kinase inhibitors with potencies comparable to or significantly higher than other known adenosine kinase inhibitors.
- the compounds are also nontoxic, particularly in connection with liver function.
- the invention concerns the compounds themselves, the preparation of these compounds, and the ]n vitro and ]n vivo adenosine kinase inhibition activity of these compounds.
- Another aspect of the invention is directed to the clinical use of the compounds to increase adenosine concentrations in biological systems.
- ]n vivo inhibition of adenosine kinase prevents phosphorylation of adenosine resulting in higher local concentrations of endogenous adenosine.
- the compounds of the invention possess advantages for pharmaceutical use such as enhanced pharmacological selectivity, efficacy, bioavailability, ease of manufacture and compound stability.
- the compounds of the invention may be used clinically to treat medical conditions where an increased localized adenosine concentration is beneficial. Accordingly, the invention is directed to the treatment of ischemic conditions such as stroke, as well as other conditions benefitted by enhanced adenosine levels, such as inflammation, arthritis, seizures, epilepsy and other neurological conditions. The compounds are also useful for treating pain, as muscle relaxants, and for inducing sleep. The invention is also directed to prodrugs and pharmaceutically acceptable salts of the compounds described, and to pharmaceutical compositions suitable for different routes of drug administration and which comprise a therapeutically effective amount of a described compound admixed with a pharmacologically acceptable carrier.
- aryl refers to aromatic groups, which have at least one ring having a conjugated pi electron system, including for example carbocyclic aryl, heterocyclic aryl and biaryl groups, all of which may be optionally substituted.
- Carbocyclic aryl groups are groups wherein all the ring atoms on the aromatic ring are carbon atoms, such as phenyl. Also included are optionally substituted phenyl groups, being preferably phenyl or phenyl substituted by one to three substituents, preferably lower alkyl, hydroxy, lower alkoxy, lower alkanoyloxy, halogen, cyano, perhalo lower alkyl, lower acylamino, lower alkoxycarbonyl, amino, alkylamino, carboxamido, and sulfamido. Further included are phenyl rings fused with a five or six membered heterocyclic aryl or carbocyclic ring, optionally containing one or more heteroatoms such as oxygen, sulfur, or nitrogen.
- Heterocyclic aryl groups are groups having from 1 to 4 heteroatoms as ring atoms in the aromatic ring and the remainder of the ring atoms carbon atoms.
- Suitable heteroatoms include oxygen, sulfur, and nitrogen, and include furanyl, thienyl, pyridyl, pyrrolyl, pyrimidyl, pyrazinyl, imidazolyl, and the like, all optionally substituted.
- Optionally substituted furanyl represents 2- or 3-furanyl or 2- or 3- furanyl preferably substituted by lower alkyl or halogen.
- Optionally substituted pyridyl represents 2-, 3- or 4-pyridyl or 2-, 3- or 4-pyridyl preferably substituted by lower alkyl or halogen.
- Optionally substituted thienyl represents 2- or 3-thienyl, or 2- or 3-thienyl preferably substituted by lower alkyl or halogen.
- biasing represents phenyl substituted by carbocyclic aryl or heterocyclic aryl as defined herein, ortho, meta or para to the point of attachment of the phenyl ring, advantageously para; biaryl is also represented as the -C 6 H 4 -Ar substituent where Ar is aryl.
- aralkyl refers to an alkyl group substituted with an aryl group. Suitable aralkyl groups include benzyl, picolyl, and the like, and may be optionally substituted.
- lower referred to herein in connection with organic radicals or compounds respectively defines such with up to and including 7, preferably up to and including 4 and advantageously one or two carbon atoms. Such groups may be straight chain or branched.
- alkyl amino refers to the groups -NRR' wherein respectively, (a) R is alkyl and R' is hydrogen, aryl or alkyl; (b) R is aryl and R' is hydrogen or aryl, and (c) R is aralkyl and R' is hydrogen or aralkyl.
- acylamino refers to RC(0)NR'.
- carbonyl refers to -C(O)-.
- acyl refers to RC(O)- where R is alkyl, aryl, aralkyl, or alkenyl.
- carboxylate refers to -CONR 2 wherein each R is independently hydrogen, lower alkyl or lower aryl.
- alkyl refers to saturated aliphatic groups including straight-chain, branched chain and cyclic groups, optionally containing one or more heteroatoms.
- alkenyl refers to unsaturated alkyl groups which contain at least one carbon-carbon double bond and includes straight-chain, branched or cyclic groups, optionally containing one or more heteroatoms such as oxygen, sulfur, or nitrogen.
- alkynyl refers to unsaturated alkyl groups which contain at least one carbon-carbon triple bond and includes straight chain, branched, or cyclic groups, optionally containing one or more heteroatoms such as oxygen, sulfur, or nitrogen.
- amino refers to -C(NH)NH 2 .
- amidoximo refers to -C(NOH)NH 2 .
- guanidino refers to -NR 1 CN(R 2 )NR 3 R 4 where R 1( R 2 , R 3 and R 4 are independently hydrogen, alkyl or aryl groups.
- aminoguanidino refers to the group -NR 1 NR 2 CN(R 3 )NR 4 R 5 where R 1( R 2 , R 3 , R 4 and R 5 are independently hydrogen, alkyl or aryl groups.
- ureido refers to the group -NR 1 C(0)NR 2 R 3 where R 1( R 2 and R 3 are independently hydrogen, alkyl or aryl groups.
- carboxylic acid refers to the group -COOH.
- acylguanidino refers to the group -C(0)NR 1 CN(R 2 )NR 3 R 4 where R ⁇ R 2 , R 3 and R 4 are independently hydrogen, alkyl or aryl groups.
- mercapto refers to SH or a tautomeric form thereof.
- alkylene refers to a divalent straight chain or branched chain saturated aliphatic radical.
- sulfonamido means -S0 2 NHR where R is hydrogen or lower alkyl.
- N-sulfonyl amine means -NHS0 2 R where R is fluoro, lower perfluoroalkyl or lower alkyl.
- N-acylated sulfonamide refers to the group -S0 2 NHCOR where R is lower alkyl or lower perfluoroalkyl.
- basic nitrogen generally refers to the nitrogen atom of an alkyl amine and implies a compound whose conjugated acid in aqueous solution has a pKa in the range of 9 to 11.
- prodrug refers to any compound that may have less intrinsic activity than the "drug” but when administered to a biological system generates the "drug” substance either as a result of spontaneous chemical reaction or by enzyme catalyzed or metabolic reaction.
- prodrugs such as acyl esters, carbonates, and urethanes, included herein as examples.
- the groups illustrated are exemplary, not exhaustive and one skilled in the art could prepare other known varieties of prodrugs. Such prodrugs fall within the scope of the invention.
- salts of compounds described herein derived from the combination of a compound of this invention and an organic or inorganic acid include salts of compounds described herein derived from the combination of a compound of this invention and an organic or inorganic acid.
- the compounds of the present invention are useful in both free base and salt form.
- a water solubilizing group is a group that increases the solubility of an inhibitor by a factor of at least 10 and preferably at least 100 at pH values suitable for intravenous administration (pH 4 to pH 10).
- salt form amounts to use of base form; both forms are within the scope of the present invention.
- treatment includes prophylactic or therapeutic administration of compounds of the invention, for the cure or amelioration of disease or symptoms associated with disease, and includes any benefit obtained or derived from the administration of the described compounds.
- the invention relates to C-4'-modified pyrrolo[2,3-d]pyrimidine and pyrazolo[3,4-d]pyrimidine nucleoside analogs of Formula 1, having activity as adenosine kinase inhibitors.
- a and B are both hydrogen, or are each independently alkenyl, the group
- n is from 1 to 4 and Q is hydrogen, hydroxy, alkyl, alkoxy, amino, azido, or halogen; or A and B together form a ring of from 3 to 6 carbons, the ring containing 0 to 3 heteroatoms selected from oxygen and nitrogen and optionally substituted by Q as defined above;
- D is halogen, aryl, aralkyl, alkyl, alkenyl, alkynyl optionally containing one or more heteroatoms such as nitrogen, oxygen or sulfur, haloalkyl, cyano, or carboxamido;
- E is nothing when Y is nitrogen; and is hydrogen, halogen, or alkyl when Y is carbon;
- F is alkyl, aryl, aralkyl, halogen, amino, alkylamino, arylamino, aralkylamino, alkoxy, aryloxy, aralkyloxy, alkylthio, arylthio, aralkylthio;
- G is hydrogen or halogen; Y is carbon or nitrogen;
- Z, and Z j> are independently hydrogen, acyl, or taken together form a cyclic carbonate; and pharmaceutically acceptable salts thereof.
- a and B are the same, but are not both methyl, and most preferably are hydrogen or (CH 2 ) n Q where n is 1 and Q is hydroxy, or amino.
- Z is preferably hydrogen, or in prodrug form is preferably acyl or carbonate ester.
- D is preferably halogen, heterocyclic aryl, phenyl or substituted phenyl;
- E is nothing when Y is nitrogen and is preferably hydrogen when Y is carbon; G is preferably hydrogen; and
- F is halogen, amino, arylamino, or heterocyclic arylamino, most preferably phenylamino or substituted phenylamino.
- Preferred substitutions are halogen, alkyl, alkoxy, or alkylamino or other groups containing a basic or acidic functionality that improves water solubility. The most preferred substitution is at the para position of phenylamino.
- prefered compounds of the invention include those where F is phenylamino, substituted at the para position with halogen (e.g. fluorine) or a water-solubilizing group.
- arylamino or phenylamino which improve water solubility have the formula (CH 2 ) r T where r is from 0 to 3 and T is an alkyl chain of 0 to 16 carbon atoms containing one or more nitrogen atoms, N-sulfonylated amino, admidoximo, N-aminoguanidino, amidino, guanidino, acyl guanidino, cyclic derivative of amidino, guanidino, or aminoguanidino, a heterocyclic aryl group, or a 5 or 6 membered alicyclic ring containing at least one basic nitrogen and optionally one or more oxygen atoms, and optionally substituted by CONW, where each V is independently an alkyl chain, at least one of which contains one or more basic nitrogen atoms, and optionally oxygen atoms, or V and V together form a six-membered ring containing at least one basic nitrogen and optionally one
- water solubilizing groups T can be an anionic group such as sulfonic acid, carboxylic acid, squaric acid derivatives, 5-tetrazolyl and other bioisosteric replacements of a carboxylic acid group such as, but not limited to, those described in Carini et al, (J. Med. Chem 34, 2525 (1991)) and references cited therein. Similar substitutions can also be made at Group D to improve water solubility.
- Prodrugs of the compounds of the present invention are included in the scope of this application. Such prodrugs may be prepared by esterification of the hydroxyl groups on the sugar ring. Specially preferred will be the ester derivatives that improve the water solubility properties.
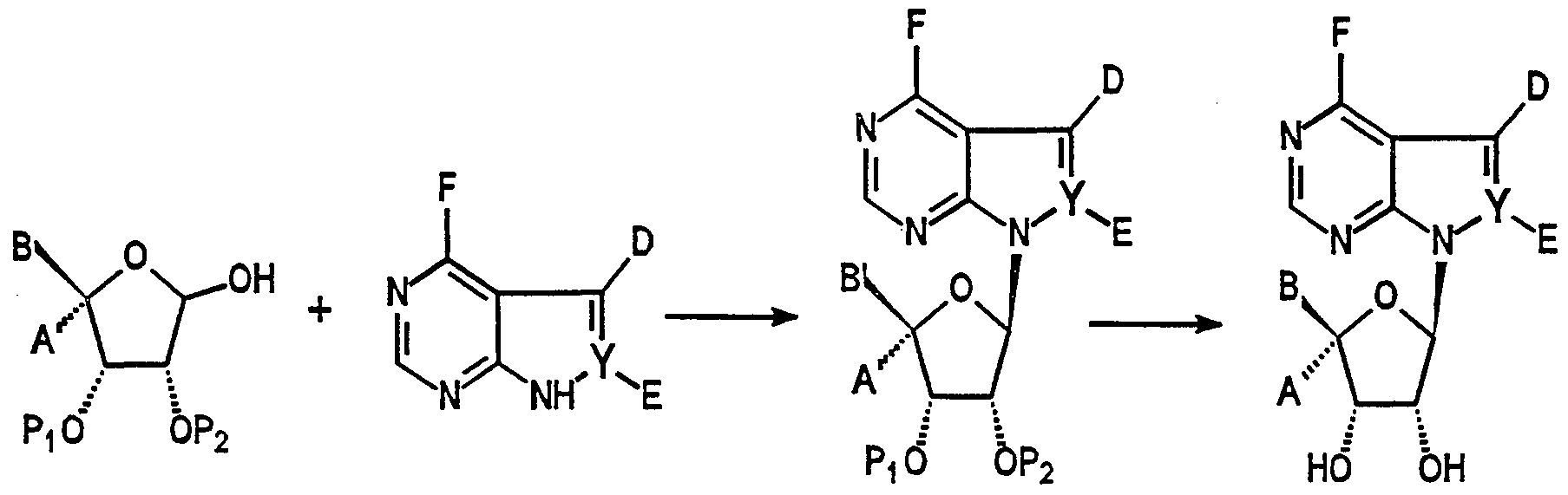
- the synthesis of compounds of the present invention can be viewed as consisting of the following steps: (A) preparation of the carbohydrate 2, (B) preparation of the heterocycle 3, (C) coupling of the carbohydrate and the heterocycle to provide a protected intermediate 4, (D) modification of substituents on the heterocycle and carbohydrate; and (E) removal of the protecting groups (Scheme 1). Each step is discussed below.
- 4-substituted carbohydrates of formula 2 are used for the synthesis of compounds of Formula 1, where A and B are both hydrogen or chosen independently from methyl, azidomethyl, aminomethyl, alkylaminomethyl, alkoxymethyl, hydroxymethyl or alkylthiomethyl.
- the formula 2 carbohydrates are made from the known methyl 2,3-O-methylethylidenefuranoside 5 (Scheme 2). See, Leonard N. J. et al. J. Heterocycl. Chem. 3, 485 (1966). The 5-alkoxy group is introduced to 5, to make 6, by the method of Snyder J. R. et al. Carbohydr. Res. 163, 169 (1987).
- the 5-deoxy, azido, amino, alkylamino, alkylthio and alternatively alkoxy carbohydrates are made by first transforming the 5-hydroxy into a leaving group L, preferably mesylate, tosylate, trifluoromethanesulfonate or halide.
- a nucleophile e.g. hydride, alkylamine, dialkylamine, alkymercaptan, alcohol or other precursors of amines such as azides or protected amines
- the isopropylidene is then replaced for less reactive protecting groups, preferably benzyl, according to methods well known to those skilled in the art. For example, Greene T. W., Protective Groups in Organic Chemistry, John Wiley & Sons, New York (1981).
- Carbohydrates for compounds of Formula 1 where A is hydroxymethyl are made by the method of Barker R. et al. J. Org. Chem. 26, 4605 (1961), to give compounds of formula 9 where A is preferably benzyl protected hydroxymethyl.
- the carbohydrate of Formula 2 is preferably prepared by the method illustrated in Scheme 3.
- the dithioacetal protecting group is removed using a modification of the procedure developed by Fetizon M. etal. J. Chem. Soc. Chem. Comm. 382 (1972), involving treatment of thioacetal with iodomethane and an inorganic base, preferably calcium carbonate.
- the second 4-C-substituent may be introduced using the procedure of Youssefyeh R. D. et al. J. Org Chem. 44(8), 1301(1979) where alkylation of an aldehyde of formula 15 with electrophile B'+ followed by a reduction gives compound 16 (Scheme 4).
- the aldehyde is obtained from the oxidative cleavage, preferably with sodium periodate, of hexofuranose 13 or oxidation of the primary alcohol of furanoside 14, preferably using a Moffat oxidation.
- Another method to obtain compounds of formula (16) is to use the procedure of Johnson C. R. et al. J. Org Chem. 59(20), 5854 (1994).
- This carbohydrate is easily obtained from erythrofuranose according to methods well known to those skilled in the art described in Greene T. W., Protective Groups in Organic Chemistry, John Wiley & Sons, New York (1981 ).
- This carbohydrate is also easily obtained by reduction of the corresponding lactone as in N. Cohen etal. J. Am. Chem.Soc. 105, 3661, (1983).
- Carbohydrates of formula 2, where A and B form a ring, e.g. cyclopropyl, are prepared from D-ribose via the well known enol ether 17 (Scheme 5). Inokawa S. et al. Carbohydr. Res. 30, 127 (1973). Cyclopropanation is performed according to the procedure of Simmons H.E. et al. J. Am Chem. Soc. 81 , 4256 (1959) or one of its many modifications. Alternatively cyclopropanation is accomplished with a diazoalkane and a metal salt, preferably palladium. Cossy J. et al. Tetrahedron Lett. 28(39), 4547 (1987). Marina ⁇ / Cyclopropanation
- Another alternative is to generate a carbene from a dihaloalkane or trihalomethane with a base in the presence of the olefin (Von E. Doering W. et al. J. Am. Chem. Soc. 76, 6162 (1954)) followed by dehalogenation, for example according to Jefford C. W. etal. J. Am. Chem. Soc. 94, 8905 (1972). Cycloaddition between diazomethane and compounds of formula 17 provides a pyrazoline intermediate which upon photolysis and deprotection produces spirocyclopropane 18 (Samano V. et al. Tetrahedron Lett. 35(21), 3445 (1994)).
- the deprotection of the anomeric center is in turn accomplished using one of the many procedures well known to those skilled in the art, e.g. Greene, T. W., Protective Groups in Organic Chemistry, John Wiley & Sons, New York, (1981).
- Carbohydrates of formula 20 are made by a wide variety of procedures. Reaction of olefin 17 with ketene under the conditions of Redlich H. et al. Angew. Chem. 101(6), 764 (1989) gives cyclobutanone 19 (Scheme 6), with is then deoxygenated using the procedure of Mori K. et al. Tetrahedron, 43(10), 2229 (1987) or Romming C. et al. Acta Chem. Scan. B , 40(6), 434 (1986). The free reducing sugar is then obtained as mentioned above (Greene T. W. Protective Groups in Organic Chemistry John Wiley & Sons, New York, 1981).
- aldehyde 15 Alkylation of aldehyde 15 with a two carbon dielectrophile, preferably diiodoethane, gives 4-disubstituted aldehyde 24 (Scheme 8, Youssefyeh R. D. et al. J. Org Chem. 44(8), 1301 (1979)).
- a metal or metal salt preferably samarium diiodide (Molander G. A. etal. J. Am. Chem. Soc. 109(2), 453 (1987)
- an organometallic reagent preferably an alkyllithium (Vanderdoes T. etal.
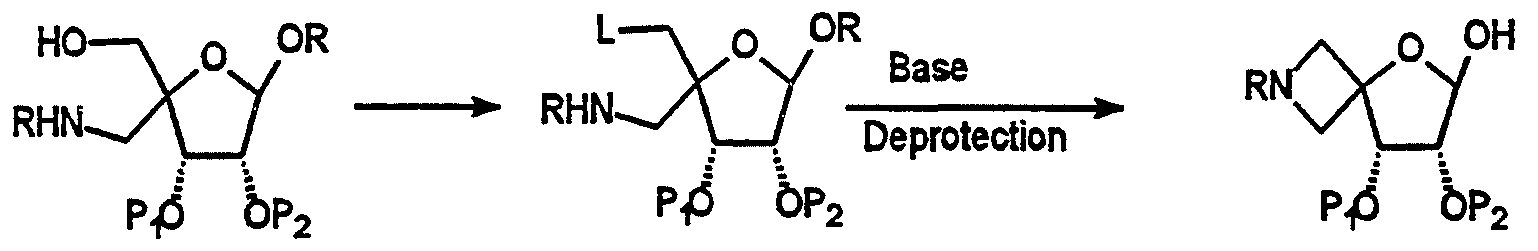
- Carbohydrate of formula 29 is made by activation of one of the primary hydroxyl groups of diol 25 (Scheme 10, Larock R. C. Comprehensive Organic Transformations, VCH Publishers, Inc. New York, (1989)). Cyclization occurs upon treatment of alcohol 28 with a base (Koll P. et al. Angew. Chem. Int. Ed. Engl. 25, 368 (1986)). The anomeric position is then deprotected as previously mentioned to afford spirooxetanofuranose 29.
- compound 29 is obtained via Mitsunobu reaction of diol 25 under the conditions of Berkowitz W. F. et al. J. Org. Chem. 52(6), 1119 (1987) which gives compound 29 after deprotection.
- lithium chloride treatment of a cyclic carbonate derived from diol 25 followed by deprotection also gives carbohydrate 29.
- 37 is made by condensing phenone 34, where L is halide or sulfonate, with phthalimide 35 in order to introduce the pyrrole nitrogen.
- Knoevenagel condensation of ketone 36 with malonitrile followed by removal of the phthalimide protecting group affords pyrrole 37.
- diaryl-pyrrolopyrimidine 39 (Taylor, E. C. et al. J. Am Chem. Soc. 87(9), 1995 (1965)). Additionally, the pyrrolopyrimidine can be further functionalized at the 6 position, when E' is methyl, by treatment with N- bromosuccinimide (Saroja ,B. etal. Tetrahedron Lett 1984, 25(47), 5429). Treatment of this bromomethylene with a nucleophile or with an alkyllithium and an electrophile allows easy introduction of functional groups such as amino or guanidino.
- the coupling of the carbohydrate 2 with pyrrolo[2,3-d]pyhmidine heterocycles is accomplished as follows (Scheme 16).
- the sugar is first converted to its 1-halo derivative, preferably chloro, by reacting it with CCI 4 and HMPT by a procedure described in Wilcox, C. T. etal. Tetrahedron Lett. 27(9), 1011 (1986).
- the halo derivative is condensed with the anion of the heterocycle 3 (where Y is carbon and E is hydrogen) using a phase transfer catalyst such as TDA-1. Rosemeyer H., and Seela, F, Helvetica Chimica Acta. 71:1573 (1988).
- the resulting blocked nucleosides are deprotected by a variety of procedures well known to those skilled in the art.
- Coupling of sugars to the pyrazzolo[3,4-d]pyrimidine bases is performed by Lewis acid catalysis conditions. Cottom, et al., J. Med. Chem.. 27, 11210 (1984). In such cases the sugars are converted to their 1 -O-acyl form, preferably 1-O-acetyl, by again using one of the many standard acetylation procedures. A mixture of the heterocycle 3 (where Y is nitrogen) and the acetylated sugar in boiling nitromethane is treated with boron trifluoride diethyl etherate. The products are purified by chromatography or crystallization, and are deprotected to obtain the final compounds.
- the final functionalization of the nucleoside is done after the coupling reaction.
- the 5-aryl group is introduced onto the pyrrolopyrimidine ring system using one of the many palladium-catalyzed cross coupling procedures (review: Stille, J. K. Ang. Chem., Int. Ed. Engl. 25, 508(1986)).
- Substitution of the aryltrialkyltin by an unsaturated trialkyi stannane by procedures as described but not limited to the one described in Stille, J. K. Ang. Chem.. Int. Ed. Engl. 25, 508 (1986) provides the 5-alkenyl derivative which can be hydrogenated in order to prepare the corresponding alkyl analog.
- guanidino derivatives can be prepared from the corresponding amino or hydroxy compounds by application of methods described in the literature such as, but not limited to, the procedures described by Miller and Bischoff (Synthesis 778 (1986)), Dodd and Kozikowski (Tetrahedron Lett. 35, 977 (1994)), Beatty and Magrath (J. Chem. Soc. 12 (1965)), Larson et al (Int. J. Pept. Protein Res. 9, 182(1977)), Brand and Brand (Org. Synth. 22, 59 (1942)), lchikawa (Tetrahedron Lett 29, 4957(1988)), Katritzky et al (Synth.
- Acid labile protecting groups such as ketals, silyl ethers or ethers are removed using a dilute acid or a weak organic acid, e.g. 0.1 N hydrochloric acid or 70% aqueous trifluoroacetic acid (Greene, T. W., Protective Groups in Organic Chemistry, John Wiley & Sons, New York (1981).
- Base labile protecting groups such as acyls or carbamates are removed by treatment with an organic or inorganic base, e.g. sodium methoxide, sodium hydroxide, ammonia (Id).
- Benzyl protecting groups are removed by hydrogenolysis in the presence of a metal catalyst, preferably palladium chloride. Shen, T. Y. et al. J. Org. Chem. 30, 835 (1965).
- Preferred compounds of the invention which can be made using the methods described, include the following.
- Hexamethylphosphorous triamide (415 ⁇ L, 1.95 mmol) was added to a solution of carbon tetrachloride (250 ⁇ L, 2.6 mmol) and the compound of Example 4 (349 mg, 0.65 mmol) in dry toluene at -78° C.
- the reaction mixture was warmed to 0°C in the course of one hour and stirred at 0°C for 30 minutes.
- the orange solution was quenched with water, diluted with toluene and washed with water and saturated aqueous sodium chloride.
- the organic layer was dried over sodium sulfate and concentrated under reduced pressure to a volume of ca. 5 mL.
- the chloro-sugar solution was added to a mixture of 4-/V-phenylamino-5-phenyl-pyrrolo[2,3- o pyrimidine (370 mg, 1.3 mmol), finely powdered potassium hydroxide (85%, 170 mg, 2.6 mmol), tris[2-(2-methoxyethoxy)ethyl]amine (420 ⁇ L, 1.3 mmol) and 4 A molecular sieves in dry toluene which had been stirring at room temperature for 2 hours. After stirring overnight at room temperature, the reaction mixture was filtered through Celite® and the filtering pad was rinsed with ethyl acetate.
- Hexamethylphosphorous triamide (800 ⁇ L, 4.35 mmol) was added to a solution of carbon tetrachloride (600 ⁇ L, 5.8 mmol) and the compound of example 11 (272 mg, 1.45 mmol) in dry toluene at -50° C.
- the reaction mixture was warmed to -10° C in the course of 30 minutes and stirred at -10° C for 15 minutes.
- the orange solution was quenched with water, diluted with toluene and washed with water and saturated aqueous sodium chloride.
- the organic layer was dried over sodium sulfate and concentrated under reduced pressure to a volume of ca. 5 mL.
- the chloro-sugar solution was added to a mixture of 4-/V-phenylamino-5-phenylpyrroIo[2,3-c pyrimidine (830 mg, 2.9 mmol), finely powdered potassium hydroxide (85%, 380 mg, 5.8 mmol) and tris[2-(2-methoxyethoxy)ethyl]amine (925 ⁇ L, 2.9 mmol) in dry toluene which had been stirring at room temperature for 90 minutes. After stirring overnight at room temperature, the reaction mixture was diluted with ethyl acetate and washed with saturated aqueous ammonium chloride. The organic layer was dried over sodium sulfate and concentrated under reduced pressure.
- the heterogeneous reaction mixture was stirred at room temperature under hydrogen (1 atm) for 6 hours, filtered through Celite ® and the filtering pad was rinsed with boiling methanol. The combined filtrates were concentrated under reduced pressure. The residue was dissolved in 0.1 N hydrochloric acid and washed twice with ethyl acetate. The pH of the aqueous solution was brought to 12 with 1 N aqueous sodium hydroxide and the resulting solution was extracted 3 times with ethyl acetate. The combined organic extracts were dried over sodium sulfate and concentrated under reduced pressure.
- the residue was purified by flash chromatography on silica gel (dichloromethane/methanol/30 % aqueous ammonium hydroxide 90/10/1 to 80/20/1).
- Example 26 Preparation of compound of formula 12 4-C-Methyl-2.3.5-tri-0-(phenylmethvn-1-(1.3-dithian-2-vn-D- ⁇ po-pentane
- a solution of the compound of Example 8 (4 g, 10 mmol) in dry tetrahydrofuran (100 mL) was added dropwise over 10 minutes to a solution of [(phenylmethyl)oxy]methyl!ithium (1.8 mmol) (Still, W. C. J. Am. Chem. Soc. 100, 1481 (1978)) in dry tetrahydrofuran (50 mL) at -78° C.
- Example 26 The title compound was synthesized following a procedure analogous to the synthesis described in Example 4.
- the reaction mixture was diluted with 0.1 N hydrochloric acid and washed twice with ethyl acetate.
- the pH of the aqueous solution was brought to 12 with 1N aqueous sodium hydroxide and the resulting solution was extracted 3 times with ethyl acetate.
- the combined organic extracts were dried over sodium sulfate and concentrated under reduced pressure.
- the residue was purified by flash chromatography on silica gel (dichloromethane/methanol/30 % aqueous ammonium hydroxide 90/10/1 to 80/20/1).
- Example 32 Preparation of compound of formula 4
- Oxalyl chloride (.55 mL, 6.3 mmol) was added dropwise, keeping the temperature below 35° C, to a solution of ⁇ /, ⁇ -dimethylformamide (4.8 mL, 63 mmol) in toluene (5.4 mL) and acetonitrile (1.9 mL).
- the slushy mixture was stirred at room temperature for 15 minutes then cooled to -12°C.
- a solution of 2,3-0- (methylethylidene)- ⁇ -D-erythrofuranose (1 g, 6.24 mmol)( Cohen, N. et al. J. Am. Chem. Soc.
- Example 63 Preparation of compound of formula 10 5-Azido-5-deoxy-2.3-di-Q-(phenylmethv ⁇ -1- ⁇ .3-dithian-2-v ⁇ -D-r/po-pentane
- the title compound was synthesized following a procedure analogous to the synthesis described in example 1.
- Example 69 Preparation of compound of formula 1 4- ⁇ /-t4-Fluorophenv ⁇ ami no-5-phenyl-7-t4-C-am inomethyl- ⁇ -D- ribofuranosvhPyrrolof2.3-d]pyrimidine dihydrochloride: Table 3 #392 lodotrimethylsilane (0.4 mL, 28 mmol) was added dropwise to a solution of 4- ⁇ /-(4-fluorophenyl)amino-5-phenyl-7-(4-C-aminomethyl-5-0-[(4- methoxyphenyl)methyl]-2,3-di-0-(phenylmethyl)- ⁇ -D-ribofuranosyl)pyrrolo[2,3- d]pyrimidine (166 mg, 0.22 mmol) in chloroform (10 mL) at 0°C.
- Triethylammonium fluoride hydrate (36 mg, 0.24 mmol) was added to a solution of 4- ⁇ /-[(1 , 1 ,2-trimethylpropyl)dimethylsilyloxymethyl]phenylamino-5-phenyl- 7-(2,3-0-(methylethylidene)- ⁇ -D-erythrofuranosyl)pyrrolo[2,3-d]pyrimidine (101 mg, 0.17 mmol) in dimethylformamide (2 mL) at rt. After stirring at rt for 30 minutes, the reaction mixture was concentrated under reduced pressure. The residue was dissolved in ethyl acetate and washed with saturated aqueous sodium chloride.
- Triphenoxyphosphonium iodide (1 g, 2.2 mmol) was added to a solution of 4- ⁇ /-(hydroxymethyl)phenylamino-5-phenyl-7-(2,3-0-(methylethylidene)- ⁇ -D- erythrofuranosyl)pyrrolo[2,3-d]pyrimidine (500 mg, 1.09 mmol) in dichloromethane (4 mL) at rt. After stirring at rt for 30 minutes, diethylamine (0.46 mL, 4.5 mmol) was added and stirring was carried on at rt overnight.
- D-erythrofuranosyl)pyrrolo[2,3-d]pyrimidine (1.2 g, 2.5 mmol), triphenylphosphine (1 g, 3.75 mmol) and phthalimide (560 mg, 3.75 mmol) in tetrahydrofuran (25 mL) at rt.
- This compound was made by a route similar to 70A, replacing phenacyl chloride by 4-fluorophenacyl chloride, followed by treatment with 4-fluoroaniline as in example 70B; m.p. 245-248 °C.
- This compound was made by a route similar to 70B by replacing 4- fluoroaniline by 4-chloroaniline.
- This compound was made by a route similar to 70B by replacing 4- fluoroaniline by 4-aminophenethyl alcohol; m.p. 206-208 °C
- Example 84 The title compound was prepared by a route similar to 70B by replacing 4- fluoroaniline by 4-aminobenzyl alcohol followed by silylation with dimethyl thexyl chlorosilane; Rf 0.5 (silica, hexanes/ethyl acetate 50/50).
- Example 84
- a and B are both HOCH 2 ; in another, A and B are both hydrogen.
- G is preferably hydrogen, and E is preferably hydrogen or bromine, most preferably hydrogen.
- Z and Z 2 are preferably hydrogen or methyl, most preferably hydrogen.
- a preferred compound is one where (107) F is 4-fluorophenylamino and D is phenyl. Using the same definitions for D, E, F, G, and Z.,, Z 2 , another preferred compound is one where (108) A is CH 3 and B is HOCH 2 .
- Still other prefered pyrolo pyrimidine compounds of the invention are those where one of A and B is CH 3 and the other is CHzNHz, or one of A and B is methoxymethyl or CH 2 OH, or one of A and B is CrJ OH and C ⁇ Nlj as shown in Table 3.
- a preferred compound is one where (147) F is 4-fluorophenylamino and D is phenyl.
- another preferred compound is one where (148) A is CH 3 and B is NH 2 CH 2 .
- a and B can together form a cyclopropyl ring.
- Prefered pyrolo compounds of this kind, where E, G, Z, and Z 2 are all hydrogen, are shown in Table 4.
- a & B Form a ring F D containing a nitrogen
- a & B Form a ring F D containing a nitrogen
- a preferred compound is one where (176) F is 4-fluorophenylamino and D is phenyl.
- Still other prefered pyrazolo pyrimidine compounds of the invention are those where one of A and B is CH 3 and the other is H 2 NCH 2 , as shown in Table 6.
- a and B can together form a cyclopropyl ring.
- Preferred pyrazolo pyrimidine nucleosides of this kind, where G, Z, and Z 2 are all hydrogen, are shown in Table 7.
- adenosine kinase inhibitors of the present invention may be used in the treatment of a variety of clinical situations where increasing local levels of adenosine are beneficial.
- the compounds of the invention act as potent inhibitors of adenosine kinase in vitro, and the present compounds in particular are orally available.
- Adenosine has been proposed to serve as a natural anticonvulsant.
- Adenosine kinase inhibitors may be used in the treatment of patients with seizures or epilepsy or patients who might have chronic low or insufficient adenosine levels or might benefit from increased adenosine such as those suffering from autism, cerebral palsy, insomnia or other neuropsychiatric symptoms.
- Adenosine kinase inhibitors of the invention find further utility in the treatment of acute pain, including but not limited to peri-operative, post-surgical, and end-stage cancer pain.
- Compounds of the invention are also useful in controlling chronic pain, including but not limited to pain caused by arthritis, cancer, trigeminal neuralgia, multiple sclerosis, neuropathies such as those arising from diabetes and AIDS and in addition, lower back pain and phantom limb pain.
- Treatment of acute and chronic pain can be treated by administration of the compounds of the invention in a systemic or oral fashion, as illustrated by animal models detailed below.
- Adenosine has been reported to be an endogenous modulator of inflammation by virtue of its effects on stimulated neutrophil function and on macrophage, lymphocyte and platelet function.
- the compounds of this invention may therefore be used in treating conditions in which inflammatory processes are prevalent such as arthritis, reperfusion injury, and other inflammatory disorders.
- the compounds of the invention are also useful in the treatment of chronic neurodegenerative disease, such as Alzheimer's disease, Parkinson's disease, ALS, Huntington's disease, and AIDS dementia.
- Stroke and central nervous system (“CNS”) trauma are conditions where tissue injury results from reduced blood supply to the CNS and are thus amenable to an intervention that provides increased levels of adenosine to the compromised tissue. It is reported that a significant component of the neurodegeneration resulting from stroke or CNS trauma is caused by increased excitatory amino acid release and sensitivity, which results in neurons being stimulated to death. In addition to vasodilatory properties, adenosine has been reported to inhibit release of excitatory amino acids (Burke and Nadler J.
- the compounds of this invention which increase adenosine levels, may also be used in the treatment of conditions where release of or sensitivity to excitatory amino acids is implicated.
- results of a series of experiments are also included. These experiments demonstrated that a number of compounds of the present invention were potent inhibitors of a purified cardiac adenosine kinase. Certain adenosine kinase inhibitors were found to inhibit seizures and exhibit anti-inflammatory activity in well-established animal models. The results of these experiments are shown in Table 8.
- Adenosine kinase activity was measured essentially as described by Yamada etal. (Yamada, Y., Goto, H., Ogasawara, N. (1988) Biochim. Biophys. Acta 660, 36-43.) with a few minor modifications. Assay mixtures contained 50 mM TRIS- maleate buffer, pH 7.0, 0.1% BSA, 1 mM ATP 1 mM MgCI 2 , 0.5 ⁇ M [U- 14 C] adenosine (400-600 mCi/mmol) and varying duplicate concentrations of inhibitor.
- the reactions were initiated by addition of approximately 0.1 ⁇ J partially purified pig heart adenosine kinase or recombinant human adenosine kinase (Spychala, J. et al., Proc.Natl.Acad.Sci. USA 93, 1232-1237, (1996)), where one unit is defined as that amount of enzyme required to phosphorylate 1 ⁇ mol adenosine per minute.
- the reactions were incubated for 20 minutes at 37° C.
- the assay was quenched upon spotting 30 ⁇ L aliquots onto 2 cm 2 pieces of Whatman DE81 anion exchange paper.
- the papersquares were washed for 3 minutes in 6 L distilled/deionized water to remove the unreacted adenosine.
- the washed squares were rinsed in 95% ethanol and dried in an oven at 100°C for 10 minutes.
- the amount of 14 C-AMP was quantified by scintillation counting.
- the concentration of inhibitor required to inhibit 50% of the adenosine kinase activity (IC ⁇ ) was determined graphically. The results for representative adenosine kinase inhibitors of the invention are shown in Table 8.
- ANTICONVULSANT ACTIVITY The anticonvulsant activity of the tested compounds was evaluated in male SA rats (100-150g, Simonsen) using the maximal electroshock (MES) model described in Swinyard et al., Antiepileptic Drugs, 3d Ed. at 85-102 (Levy, et al., eds.), NY: Raven Press (1989).
- the rats were maintained on a 12/12 light/dark cycle in temperature controlled facilities with free access to food and water.
- the animals are fasted overnight, prior to the experiment.
- One hour prior to seizure testing the animals were injected interperitoneally (ip) or orally (per os, po) with one of various doses of test compound dissolved in DMSO or PEG 400.
- MES Maximal electroshock seizures
- HTE hind limb tonic extension
- the effective dose at which 50% of the rats were protected was calculated from a dose response curve.
- the results for exemplary compounds of the invention are set forth in Table 8, expressed as ED ⁇ values. For compounds where the ED ⁇ was not calculated, the result is listed as >5 if HTE was inhibited in fewer than 50% of the animals in the intial screen, or ⁇ 5 if HTE was inhibited in more than 50% of the animals in the intial screen.
- Carrageenan (Type ⁇ ) was suspended in sterile PBS at 1% (w/v), autoclaved for 30 minutes, and stored at room temperature. Rats were pretreated with vehicle or AK inhibitor (10 mg/kg) by oral gavage or i.p. administration and the volume of the left hind paw was measured using a water displacement plethysmometer (Stoelting Co., Wood Dale, IL). One hour after oral treatment or 30 minutes after i.p. treatment, the rats were briefly anaesthetized, and 0.1 ml of the carrageenan solution was injected subcutaneously into the planar surface of the left hind paw. The ensuing paw swelling was measured by plethysmometry after 3 hours.
- Serum was prepared and liver enzymes (serum glutamic-oxaloacetic transaminase (SGOT), serum glutamic-pyruvic transaminase (SGPT)) and total bilirubin in the serum samples were determined by a commercial laboratory. Results are shown in table 9.
- Rat skin lesions were induced as in Rosengren et al., J. Immunology 154: 5444-51 (1995).
- the dorsal skin of male SA rats was shaved and carrageenan (Type ⁇ ) or phosphate-buffered saline was injected intradermally. Three hours later, the injection sites were biopsied and weighed. Neutrophil content of the skin biopsies was measured as the quantity of myeloperoxidase (MPO) present in a tissue homogenate.
- MPO myeloperoxidase
- the excised, weighed skin pieces were placed in 4 ml 0.5% mixed alkyl trimethylammonium bromide and homogenized at the highest speed for 15 seconds in a Polytron homogenizer (Brinkmann Instruments, Westbury, NY).
- Lipids were extracted by adding 1 ml of dichloromethane to the homogenate, vortexing vigorously, and centrifuging at lOOOg, 5°C for 15 min. Fifty ⁇ of each supernatant was added in duplicate to a 96 well assay plate, along with dilutions of human myeloperoxidase standard. Potassium phosphate buffer (pH 6.1) containing 0.36 mg/ml of ⁇ -dianisidine dihydrochloride and 0.001% hydrogen peroxide was added (200 ⁇ l/well), and the absorbance at 450 nm was read after 5 min incubation at room temperature. The myeloperoxidase content of each skin piece was calculated from a standard curve constructed using least-square regression, and expressed as units of MPO/g of tissue.
- AK inhibitors were administered orally using polyethylene glycol-400 as vehicle or intraperitoneally using dimethylsulfoxide as vehicle, at indicated time before skin lesion injection. For each experiment, the average of all values from saline-induced lesions was calculated. This baseline value was then subtracted from values obtained from carrageenan-induced lesions. Percent inhibition for AK inhibitors were calculated from these baseline-corrected values. The results are shown in Table 10.
- Heat-killed Mycobacterium butyricu was ground to a find powder and suspended in heavy mineral oil at 10 mg/ml. The suspension was injected subcutaneously into the base of the tail of male Lewis rats at 0.1 ml per rat. This immunization procedure induces an agressive arthritis that is apparent at day 10-12 and rapidly worsens. The volumes of the hind paws were measured before immunization and on days 12, 15 and 20 after immunization. The baseline paw volume was subtracted from the arthritic volumes to yield paw swelling. AK inhibitors were given by daily oral gavage beginning on day 4 after immunization, using polyethylene glycol-400 as the vehicle. Control rats received vehicle only. Percent inhibition was calculated based on paw swelling in AK inhibitor treated group compared to vehicle treated group, and is reported in Table 11.
- Phase 1 of the response which is brief, lasting approximately 0 - 5 min post-injection, is followed by a more prolonged phase 2, lasting approximately 10 - 80 min post-injection.
- Phase 1 behavior is thought to be a direct effect of the irritant on nociceptors at the injection site while phase 2 behavior is thought to include a hyperalgesic component mediated by sensitization of neuronal elements within the spinal cord.
- Phase 2a Phase 2a
- Rats Male, Simonsen weighing between 100 - 200 g, are used in the present experiments.
- drugs are administered orally 90 min prior to initiation of formalin test.
- the animals in groups of 4 are placed individually in a small animal restrainer with the right hindpaw accessible through a hole in the bottom of the restrainer.
- the formalin paw assay is initiated by the injection using a 30G needle of 50 ⁇ l of a 5% formalin solution in saline into the right plantar surface of each hindpaw.
- the rat is then immediately placed in a separate plexiglass box and scoring (described below) of the animal's behavior is begun at 1.7 min after formalin injection.
- the instantaneous behavior of each animal in a group of 4 was observed and assigned a score once in each 20 second interval. This sequence is repeated over a 30 min period.
- the scoring protocol is an adaptation of the method published by Dubuisson and Dennis (Pain 4:161 -174, 1977) which assigns a score from 0 - 3 as follows:
- 0.1 to 200 nmole/min/kg preferably from 1 to 50 nmol/min/kg.
- Such rates are easily maintained when soluble compounds are intravenously administered as discussed below.
- use of time-release preparations to control the rate of release of the active ingredient may be preferred.
- These compounds are administered in a dose of about 0.01 mg/kg/day to about 100 mg/kg/day, preferably from about 0.1 mg/kg/day to about 10 mg/kg/day.
- the compounds of the invention may be administered by a variety of means including orally, parenterally, by inhalation spray, topically, or rectally in formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, and intraarterial injections with a variety of infusion techniques.
- Intraarterial and intravenous injection as used herein includes administration through catheters. Preferred for certain indications are methods of administration which allow rapid access to the tissue or organ being treated, such as intravenous injections for the treatment of myocardial infarction. When an organ outside a body is being treated, perfusion is preferred.
- compositions containing the active ingredient may be in any form suitable for the intended method of administration.
- tablets, troches, lozenges, aqueous or oil suspensions, dispersible powders or granules, emulsions, hard or soft capsules, syrups or elixirs may be prepared.
- Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents including those from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide a palatable preparation.
- Tablets containing the active ingredient in admixture with non-toxic pharmaceutically acceptable excipient which are suitable for manufacture of tablets are acceptable.
- excipients may be, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, such as maize starch, or alginic acid; binding agents, such as starch, gelatin or acacia; and lubricating agents, such as magnesium stearate, stearic acid or talc. Tablets may be uncoated or may be coated by known techniques including microencapsulation to delay disintegration and adsorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate alone or with a wax may be employed.
- inert diluents such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate
- granulating and disintegrating agents such as maize starch, or alginic acid
- Formulations for oral use may be also presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, such as peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example calcium phosphate or kaolin
- an oil medium such as peanut oil, liquid paraffin or olive oil.
- Aqueous suspensions of the invention contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients include a suspending agent, such as sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethylcelluose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia, and dispersing or wetting agents such as a naturally occurring phosphatide (e.g., lecithin), a condensation product of an alkylene oxide with a fatty acid (e.g., polyoxyethylene stearate), a condensation product of ethylene oxide with a long chain aliphatic alcohol (e.g., heptadeaethyleneoxycetanol), a condensation product of ethylene oxide with a partial ester derived from a fatty acid and a hexitol anhydride (e.g., polyoxyethylene sorbitan mono-oleate).
- a suspending agent
- the aqueous suspension may also contain one or more preservative such as ethyl of n-propyl p- hydroxybenzoate, one or more coloring agent, one or more flavoring agent and one or more sweetening agent, such as sucrose or saccharin.
- preservative such as ethyl of n-propyl p- hydroxybenzoate
- coloring agent such as a coloring agent
- flavoring agent such as sucrose or saccharin.
- sweetening agent such as sucrose or saccharin.
- Oil suspensions may be formulated by suspending the active ingredient in a vegetable oil, such as arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
- the oral suspensions may contain a thickening agent, such as beeswax, hard paraffin or cetyl alcohol.
- Sweetening agents, such as those set forth above, and flavoring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an antioxidant such as ascorbic acid.
- Dispersible powders and granules of the invention suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, a suspending agent, and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those disclosed above. Additional excipients, for example sweetening, flavoring and coloring agents, may also be present.
- the pharmaceutical compositions of the invention may also be in the form of oil-in-water emulsions.
- the oily phase may be a vegetable oil, such as olive oil or arachis oil, a mineral oil, such as liquid paraffin, or a mixture of these.
- Suitable emulsifying agents include naturally-occurring gums, such as gum acacia and gum tragacanth, naturally occurring phosphatides, such as soybean lecithin, esters or partial esters derived from fatty acids and hexitol anhydrides, such as sorbitan mono- oleate, and condensation products of these partial esters with ethylene oxide, such as polyoxyethylene sorbitan mono-oleate.
- the emulsion may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, such as glycerol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, a flavoring or a coloring agent.
- the pharmaceutical compositions of the invention may be in the form of a sterile injectable preparation, such as a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, such as a solution in 1,3-butanediol or prepared as a lyophylized powder.
- a non-toxic parenterally-acceptable diluent or solvent such as a solution in 1,3-butanediol or prepared as a lyophylized powder.
- sterile fixed oils may conventionally be employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid may likewise be used in the preparation of injectables.
- a time-release formulation intended for oral administration to humans may contain 20 to 1000 ⁇ moles of active material compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95% of the total compositions. It is preferred that pharmaceutical composition be prepared which provides easily measurable amounts for administration.
- an aqueous solution intended for intravenous infusion should contain from about 0.1 to about 15 ⁇ moles of the active ingredient per ML of solution so that infusion of a suitable volume at a rate of about 30 mL/hr can occur.
- formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion.
- the active ingredient may also be administered as a bolus, electuary or paste.
- a tablet may be made by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder (e.g.. povidone, gelatin, hydroxypropylmethyl cellulose), lubricant, inert diluent, preservative, disintegrant (e.g.. sodium starch glycolate, cross-linked povidone, cross-linked sodium carboxymethyl cellulose) surface-active or dispersing agent.
- Molded tablets may be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- the tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein using, for example, hydroxypropylmethyl cellulose in varying proportions to provide the desired release profile. Tablets may optionally be provided with an enteric coating, to provide release in parts of the gut other than the stomach. This is particularly advantageous with the compounds of formula (I) as such compounds are susceptible to acid hydrolysis.
- Formulations suitable for topical administration in the mouth include lozenges comprising the active ingredient in a flavored basis, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert basis such as gelatin and glycerin, or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- Formulations for rectal administration may be presented as a suppository with a suitable base comprising for example cocoa butter or a salicylate.
- Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations containing in addition to the ddPN ingredient such carriers as are known in the art to be appropriate.
- Formations suitable for parenteral administration include aqueous and non-aqueous isotonic sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the formulations may be presented in unit-dose or multi-dose sealed containers, for example, ampoules and vials, and may be sorted in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- sterile liquid carrier for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- Preferred unit dosage formulations are those containing a daily dose or unit, daily sub-dose, or an appropriate fraction thereof, of an adenosine kinase inhibitor compound. It will be understood, however, that the specific dose level for any particular patient will depend on a variety of factors including the activity of the specific compound employed; the age, body weight, general health, sex and diet of the individual being treated; the time and route of administration; the rate of excretion; other drugs which have previously been administered; and the severity of the particular disease undergoing therapy, as is well understood by those skilled in the art.
- Capsules comprising adenosine kinase inhibitors suitable for oral administration according to the methods of the present invention may be prepared as follows: (1) for a 10,000 capsule preparation: 1500 g of adenosine kinase inhibitor is blended with other ingredients (as described above) and filled into capsules which are suitable for administration depending on dose, from about 4 capsules per day (1 per 6 hours) to about 8 capsules per day (2 capsules per 6 hours), to an adult human.
Abstract
Description




























Claims

Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT96919442T ATE257841T1 (en) | 1995-06-07 | 1996-06-07 | C-4' MODIFIED ADENOSIN KINASE INHIBITORS |
| BR9609013-8A BR9609013A (en) | 1995-06-07 | 1996-06-07 | Nucleoside analog, and, compound. |
| JP9502290A JPH11507387A (en) | 1995-06-07 | 1996-06-07 | C-4 'modified adenosine kinase inhibitor |
| EA199800009A EA199800009A1 (en) | 1995-06-07 | 1996-06-07 | Adenosynkinase inhibitors, modified by the C-4 ′ atom |
| APAP/P/1997/001165A AP9701165A0 (en) | 1995-06-07 | 1996-06-07 | C-4' adnosine kinase inhibitors. |
| AU61783/96A AU6178396A (en) | 1995-06-07 | 1996-06-07 | C-4' modified adenosine kinase inhibitors |
| EP96919442A EP0832091B1 (en) | 1995-06-07 | 1996-06-07 | C-4' modified adenosine kinase inhibitors |
| IL12233596A IL122335A0 (en) | 1995-06-07 | 1996-06-07 | C-4' adenosine kinase inhibitors |
| DE69631330T DE69631330T2 (en) | 1995-06-07 | 1996-06-07 | C-4 'MODIFIED ADENOSINE KINAS INHIBITORS |
| SK1660-97A SK166097A3 (en) | 1995-06-07 | 1996-06-07 | C-4'-modified adenosine kinase inhibitors |
| IS4621A IS4621A (en) | 1995-06-07 | 1997-11-26 | C-4 ′ Transformed adenosine kinase inhibitors |
| NO975585A NO975585L (en) | 1995-06-07 | 1997-12-03 | C-4 'modified adenosine kinase inhibitors |
| BG102163A BG102163A (en) | 1995-06-07 | 1998-01-07 | Inhibitors of adenosinekinase modified in c-4'position |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/486,161 | 1995-06-07 | ||
| US08/486,161 US5674998A (en) | 1989-09-15 | 1995-06-07 | C-4' modified adenosine kinase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1996040705A1 true WO1996040705A1 (en) | 1996-12-19 |
Family
ID=23930844
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1996/010404 WO1996040705A1 (en) | 1995-06-07 | 1996-06-07 | C-4' modified adenosine kinase inhibitors |
Country Status (24)
| Country | Link |
|---|---|
| US (1) | US5674998A (en) |
| EP (1) | EP0832091B1 (en) |
| JP (1) | JPH11507387A (en) |
| KR (1) | KR19990022740A (en) |
| CN (1) | CN1190401A (en) |
| AP (1) | AP9701165A0 (en) |
| AT (1) | ATE257841T1 (en) |
| AU (1) | AU6178396A (en) |
| BG (1) | BG102163A (en) |
| BR (1) | BR9609013A (en) |
| CA (1) | CA2220642A1 (en) |
| CZ (1) | CZ392797A3 (en) |
| DE (1) | DE69631330T2 (en) |
| EA (1) | EA199800009A1 (en) |
| HU (1) | HUP9802193A3 (en) |
| IL (1) | IL122335A0 (en) |
| IS (1) | IS4621A (en) |
| MX (1) | MX9709859A (en) |
| NO (1) | NO975585L (en) |
| OA (1) | OA10639A (en) |
| PL (1) | PL323904A1 (en) |
| SK (1) | SK166097A3 (en) |
| TR (1) | TR199701539T1 (en) |
| WO (1) | WO1996040705A1 (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6271370B1 (en) | 1999-05-11 | 2001-08-07 | Pfizer Inc | Process for the synthesis of nucleoside analogs |
| WO2001058920A2 (en) * | 2000-02-10 | 2001-08-16 | Mitsui Chemicals, Inc. | Process for selectively producing 1-phosphorylated sugar derivative anomer and process for producing nucleoside |
| US6660744B1 (en) | 1999-09-17 | 2003-12-09 | Abbott Gmbh & Co. Kg | Pyrazolopyrimidines as therapeutic agents |
| WO2004022572A1 (en) * | 2002-09-06 | 2004-03-18 | Alchemia Limited | Compounds that interact with kinases |
| US6713474B2 (en) | 1998-09-18 | 2004-03-30 | Abbott Gmbh & Co. Kg | Pyrrolopyrimidines as therapeutic agents |
| US7071199B1 (en) | 1999-09-17 | 2006-07-04 | Abbott Gmbh & Cco. Kg | Kinase inhibitors as therapeutic agents |
| US7863444B2 (en) | 1997-03-19 | 2011-01-04 | Abbott Laboratories | 4-aminopyrrolopyrimidines as kinase inhibitors |
| WO2011005860A3 (en) * | 2009-07-07 | 2013-05-10 | Alnylam Pharmaceuticals, Inc. | 5' phosphate mimics |
| US9073960B2 (en) | 2011-12-22 | 2015-07-07 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9156845B2 (en) | 2012-06-29 | 2015-10-13 | Pfizer Inc. | 4-(substituted amino)-7H-pyrrolo[2,3-d] pyrimidines as LRRK2 inhibitors |
| WO2016066582A1 (en) | 2014-10-28 | 2016-05-06 | Bci Pharma | Nucleoside kinase inhibitors |
| US9422322B2 (en) | 2013-06-26 | 2016-08-23 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9441007B2 (en) | 2012-03-21 | 2016-09-13 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9695171B2 (en) | 2013-12-17 | 2017-07-04 | Pfizer Inc. | 3,4-disubstituted-1 H-pyrrolo[2,3-b]pyridines and 4,5-disubstituted-7H-pyrrolo[2,3-c]pyridazines as LRRK2 inhibitors |
| US9862743B2 (en) | 2013-10-11 | 2018-01-09 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10039753B2 (en) | 2015-09-14 | 2018-08-07 | Pfizer Inc. | Imidazo[4,5-c]quinoline and imidazo[4,5-c][1,5]naphthyridine derivatives as LRRK2 inhibitors |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| WO2021023888A1 (en) | 2019-08-08 | 2021-02-11 | B.C.I. Pharma | Isoquinoline derivatives as protein kinase inhibitors |
| US11697666B2 (en) | 2021-04-16 | 2023-07-11 | Gilead Sciences, Inc. | Methods of preparing carbanucleosides using amides |
| US11767337B2 (en) | 2020-02-18 | 2023-09-26 | Gilead Sciences, Inc. | Antiviral compounds |
Families Citing this family (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE159257T1 (en) * | 1994-05-03 | 1997-11-15 | Ciba Geigy Ag | PYRROLOPYRIMIDE DERIVATIVES WITH ANTIPROLIFERATIVE EFFECT |
| CA2224435C (en) * | 1995-07-06 | 2008-08-05 | Novartis Ag | Pyrrolopyrimidines and processes for the preparation thereof |
| US6051578A (en) * | 1996-02-12 | 2000-04-18 | Pfizer Inc. | Pyrazolopyrimidines for treatment of CNS disorders |
| US6831069B2 (en) | 1999-08-27 | 2004-12-14 | Ribapharm Inc. | Pyrrolo[2,3-d]pyrimidine nucleoside analogs |
| WO2001027114A1 (en) * | 1999-08-27 | 2001-04-19 | Icn Pharmaceuticals, Inc. | PYRROLO[2,3-d]PYRIMIDINE NUCLEOSIDE ANALOGS |
| WO2001043731A2 (en) * | 1999-12-16 | 2001-06-21 | Alcon, Inc. | Inhibitors of adenosine kinase for the treatment of optic nerve and retinal damage |
| US7638496B2 (en) | 2000-02-15 | 2009-12-29 | Valeant Pharmaceuticals North America | Nucleoside analogs with carboxamidine modified monocyclic base |
| US7414036B2 (en) * | 2002-01-25 | 2008-08-19 | Muscagen Limited | Compounds useful as A3 adenosine receptor agonists |
| BRPI0400869B8 (en) * | 2004-03-02 | 2021-05-25 | Univ Estadual Campinas Unicamp | new compounds derived from 4-anilinoquinazolines with adenosine kinase inhibiting property |
| US9512125B2 (en) | 2004-11-19 | 2016-12-06 | The Regents Of The University Of California | Substituted pyrazolo[3.4-D] pyrimidines as anti-inflammatory agents |
| RS52458B (en) | 2005-02-04 | 2013-02-28 | Millennium Pharmaceuticals Inc. | Inhibitors of e1 activating enzymes |
| EP1915053A2 (en) * | 2005-08-12 | 2008-04-30 | Merck & Co., Inc. | Novel 2'-c-methyl and 4'-c-methyl nucleoside derivatives |
| EP1989206B1 (en) | 2006-02-02 | 2012-07-04 | Millennium Pharmaceuticals, Inc. | Inhibitors of e1 activating enzyme |
| MX2008012928A (en) | 2006-04-04 | 2009-03-06 | Univ California | P13 kinase antagonists. |
| WO2009046448A1 (en) | 2007-10-04 | 2009-04-09 | Intellikine, Inc. | Chemical entities and therapeutic uses thereof |
| KR101897881B1 (en) | 2008-01-04 | 2018-09-12 | 인텔리카인, 엘엘씨 | Certain chemical entities, compositions and methods |
| US8193182B2 (en) * | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| EP2252293B1 (en) | 2008-03-14 | 2018-06-27 | Intellikine, LLC | Kinase inhibitors and methods of use |
| US8993580B2 (en) | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| EP3009436B1 (en) | 2008-07-08 | 2019-06-05 | Intellikine, LLC | Kinase inhibitors and methods of use |
| WO2010006072A2 (en) | 2008-07-08 | 2010-01-14 | The Regents Of The University Of California | Mtor modulators and uses thereof |
| JP5731978B2 (en) | 2008-09-26 | 2015-06-10 | インテリカイン, エルエルシー | Heterocyclic kinase inhibitor |
| EP2358720B1 (en) | 2008-10-16 | 2016-03-02 | The Regents of The University of California | Fused ring heteroaryl kinase inhibitors |
| US8476282B2 (en) | 2008-11-03 | 2013-07-02 | Intellikine Llc | Benzoxazole kinase inhibitors and methods of use |
| CA2760791C (en) | 2009-05-07 | 2017-06-20 | Intellikine, Inc. | Heterocyclic compounds and uses thereof |
| WO2010132110A1 (en) | 2009-05-14 | 2010-11-18 | Millennium Pharmaceuticals, Inc. | Hydrochloride salt of ((1s,2s,4r)-4-{4-[(1s)-2,3-dihydro-1h-inden-1-ylamino]-7h-pyrrolo [2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate |
| US8980899B2 (en) | 2009-10-16 | 2015-03-17 | The Regents Of The University Of California | Methods of inhibiting Ire1 |
| CN103002738A (en) | 2010-05-21 | 2013-03-27 | 英特利凯恩有限责任公司 | Chemical compounds, compositions and methods for kinase modulation |
| WO2012064973A2 (en) | 2010-11-10 | 2012-05-18 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| UA115767C2 (en) | 2011-01-10 | 2017-12-26 | Інфініті Фармасьютікалз, Інк. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| CN106619647A (en) | 2011-02-23 | 2017-05-10 | 因特利凯有限责任公司 | Combination of mtor inhibitors and pi3-kinase inhibitors and uses thereof |
| US8969363B2 (en) | 2011-07-19 | 2015-03-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| AU2012284091B2 (en) | 2011-07-19 | 2015-11-12 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| WO2013032591A1 (en) | 2011-08-29 | 2013-03-07 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| EP2751112B1 (en) | 2011-09-02 | 2019-10-09 | The Regents of The University of California | Substituted pyrazolo[3,4-d]pyrimidines and uses thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| BR112015006828A8 (en) | 2012-09-26 | 2019-09-17 | Univ California | compound, or a pharmaceutically acceptable salt thereof; pharmaceutical composition; use of the compound; and method for modulating the activity of an ire1 protein |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| TWI657085B (en) | 2013-10-04 | 2019-04-21 | 英菲尼提製藥股份有限公司 | Heterocyclic compounds and uses thereof |
| JP6701088B2 (en) | 2014-03-19 | 2020-05-27 | インフィニティー ファーマシューティカルズ, インコーポレイテッド | Heterocyclic compounds for use in the treatment of PI3K-gamma mediated disorders |
| WO2015160975A2 (en) | 2014-04-16 | 2015-10-22 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| WO2017024310A1 (en) | 2015-08-06 | 2017-02-09 | Chimerix, Inc. | Pyrrolopyrimidine nucleosides and analogs thereof useful as antiviral agents |
| KR20180058741A (en) | 2015-09-14 | 2018-06-01 | 인피니티 파마슈티칼스, 인코포레이티드 | Solid form of isoquinolines, a process for their preparation, compositions comprising them and methods for using them |
| WO2017161116A1 (en) | 2016-03-17 | 2017-09-21 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as pi3k kinase inhibitors |
| WO2017214269A1 (en) | 2016-06-08 | 2017-12-14 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| EP3474856B1 (en) | 2016-06-24 | 2022-09-14 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US11111264B2 (en) | 2017-09-21 | 2021-09-07 | Chimerix, Inc. | Morphic forms of 4-amino-7-(3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide and uses thereof |
| KR102639275B1 (en) * | 2021-06-08 | 2024-02-21 | 퓨쳐메디신 주식회사 | Nucleoside derivative having kinase multiple target inhibiting activity and pharmaceutical composition for preventing and treating cancer comprising the same |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4904666A (en) * | 1987-04-15 | 1990-02-27 | Boehringer Mannheim Gmbh | Pyrazolo(3,4-d)pyrimidine compounds, compositions and method of use |
| EP0496617A1 (en) * | 1991-01-23 | 1992-07-29 | Gensia, Inc. | Adenosine kinase inhibitors |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4455420A (en) * | 1983-01-13 | 1984-06-19 | Hoffmann-La Roche Inc. | 4-Amino-7-(5-deoxy-beta-D-ribofuranosyl)-5-iodo-7H-pyrrolo[2,3-d] pyrimidine |
| AU665184B2 (en) * | 1991-01-23 | 1995-12-21 | Gensia, Inc. | Adenosine kinase inhibitors |
| IL108523A0 (en) * | 1993-02-03 | 1994-05-30 | Gensia Inc | Pharmaceutical compositions containing adenosine kinase inhibitors for preventing or treating conditions involving inflammatory responses and pain |
| JPH08506343A (en) * | 1993-02-03 | 1996-07-09 | ジェンシア・インコーポレイテッド | Adenosine kinase inhibitors containing lysofuranosyl derivatives |
| US6143749A (en) * | 1995-06-07 | 2000-11-07 | Abbott Laboratories | Heterocyclic substituted cyclopentane compounds |
-
1995
- 1995-06-07 US US08/486,161 patent/US5674998A/en not_active Expired - Fee Related
-
1996
- 1996-06-07 AU AU61783/96A patent/AU6178396A/en not_active Abandoned
- 1996-06-07 DE DE69631330T patent/DE69631330T2/en not_active Expired - Fee Related
- 1996-06-07 HU HU9802193A patent/HUP9802193A3/en unknown
- 1996-06-07 PL PL96323904A patent/PL323904A1/en unknown
- 1996-06-07 IL IL12233596A patent/IL122335A0/en unknown
- 1996-06-07 TR TR97/01539T patent/TR199701539T1/en unknown
- 1996-06-07 CA CA002220642A patent/CA2220642A1/en not_active Abandoned
- 1996-06-07 AP APAP/P/1997/001165A patent/AP9701165A0/en unknown
- 1996-06-07 CN CN96195383A patent/CN1190401A/en active Pending
- 1996-06-07 CZ CZ973927A patent/CZ392797A3/en unknown
- 1996-06-07 SK SK1660-97A patent/SK166097A3/en unknown
- 1996-06-07 JP JP9502290A patent/JPH11507387A/en not_active Ceased
- 1996-06-07 BR BR9609013-8A patent/BR9609013A/en unknown
- 1996-06-07 EA EA199800009A patent/EA199800009A1/en unknown
- 1996-06-07 WO PCT/US1996/010404 patent/WO1996040705A1/en active IP Right Grant
- 1996-06-07 KR KR1019970709182A patent/KR19990022740A/en not_active Application Discontinuation
- 1996-06-07 AT AT96919442T patent/ATE257841T1/en not_active IP Right Cessation
- 1996-06-07 EP EP96919442A patent/EP0832091B1/en not_active Expired - Lifetime
-
1997
- 1997-11-26 IS IS4621A patent/IS4621A/en unknown
- 1997-12-03 OA OA70150A patent/OA10639A/en unknown
- 1997-12-03 NO NO975585A patent/NO975585L/en unknown
- 1997-12-08 MX MX9709859A patent/MX9709859A/en unknown
-
1998
- 1998-01-07 BG BG102163A patent/BG102163A/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4904666A (en) * | 1987-04-15 | 1990-02-27 | Boehringer Mannheim Gmbh | Pyrazolo(3,4-d)pyrimidine compounds, compositions and method of use |
| EP0496617A1 (en) * | 1991-01-23 | 1992-07-29 | Gensia, Inc. | Adenosine kinase inhibitors |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7863444B2 (en) | 1997-03-19 | 2011-01-04 | Abbott Laboratories | 4-aminopyrrolopyrimidines as kinase inhibitors |
| US6713474B2 (en) | 1998-09-18 | 2004-03-30 | Abbott Gmbh & Co. Kg | Pyrrolopyrimidines as therapeutic agents |
| US6271370B1 (en) | 1999-05-11 | 2001-08-07 | Pfizer Inc | Process for the synthesis of nucleoside analogs |
| US6660744B1 (en) | 1999-09-17 | 2003-12-09 | Abbott Gmbh & Co. Kg | Pyrazolopyrimidines as therapeutic agents |
| US7071199B1 (en) | 1999-09-17 | 2006-07-04 | Abbott Gmbh & Cco. Kg | Kinase inhibitors as therapeutic agents |
| WO2001058920A2 (en) * | 2000-02-10 | 2001-08-16 | Mitsui Chemicals, Inc. | Process for selectively producing 1-phosphorylated sugar derivative anomer and process for producing nucleoside |
| WO2001058920A3 (en) * | 2000-02-10 | 2001-11-08 | Mitsui Chemicals Inc | Process for selectively producing 1-phosphorylated sugar derivative anomer and process for producing nucleoside |
| KR100517163B1 (en) * | 2000-02-10 | 2005-09-26 | 미쯔이카가쿠 가부시기가이샤 | Process for selectively producing 1-phosphorylated sugar derivative anomer and process for producing nucleoside |
| US7038039B2 (en) | 2000-02-10 | 2006-05-02 | Mitsui Chemicals, Inc. | Process for selectively producing 1-phosphorylated sugar derivative anomer and process for producing nucleoside |
| US8673586B2 (en) | 2002-09-06 | 2014-03-18 | Alchemia Limited | Compounds that interact with kinases |
| WO2004022572A1 (en) * | 2002-09-06 | 2004-03-18 | Alchemia Limited | Compounds that interact with kinases |
| US7291623B2 (en) | 2002-09-06 | 2007-11-06 | Alchemia Limited | Compounds that interact with kinases |
| US10131908B2 (en) | 2009-07-07 | 2018-11-20 | Alnylam Pharmaceuticals, Inc. | 5′ phosphate mimics |
| WO2011005860A3 (en) * | 2009-07-07 | 2013-05-10 | Alnylam Pharmaceuticals, Inc. | 5' phosphate mimics |
| US8927513B2 (en) | 2009-07-07 | 2015-01-06 | Alnylam Pharmaceuticals, Inc. | 5′ phosphate mimics |
| US9073960B2 (en) | 2011-12-22 | 2015-07-07 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US11021509B2 (en) | 2011-12-22 | 2021-06-01 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10464965B2 (en) | 2011-12-22 | 2019-11-05 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9441007B2 (en) | 2012-03-21 | 2016-09-13 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10485815B2 (en) | 2012-03-21 | 2019-11-26 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9156845B2 (en) | 2012-06-29 | 2015-10-13 | Pfizer Inc. | 4-(substituted amino)-7H-pyrrolo[2,3-d] pyrimidines as LRRK2 inhibitors |
| US9642855B2 (en) | 2012-06-29 | 2017-05-09 | Pfizer Inc. | Substituted pyrrolo[2,3-d]pyrimidines as LRRK2 inhibitors |
| US10696708B2 (en) | 2013-06-26 | 2020-06-30 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9932363B2 (en) | 2013-06-26 | 2018-04-03 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9422322B2 (en) | 2013-06-26 | 2016-08-23 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10370401B2 (en) | 2013-10-11 | 2019-08-06 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9862743B2 (en) | 2013-10-11 | 2018-01-09 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9695171B2 (en) | 2013-12-17 | 2017-07-04 | Pfizer Inc. | 3,4-disubstituted-1 H-pyrrolo[2,3-b]pyridines and 4,5-disubstituted-7H-pyrrolo[2,3-c]pyridazines as LRRK2 inhibitors |
| WO2016066582A1 (en) | 2014-10-28 | 2016-05-06 | Bci Pharma | Nucleoside kinase inhibitors |
| US10039753B2 (en) | 2015-09-14 | 2018-08-07 | Pfizer Inc. | Imidazo[4,5-c]quinoline and imidazo[4,5-c][1,5]naphthyridine derivatives as LRRK2 inhibitors |
| WO2021023888A1 (en) | 2019-08-08 | 2021-02-11 | B.C.I. Pharma | Isoquinoline derivatives as protein kinase inhibitors |
| US11767337B2 (en) | 2020-02-18 | 2023-09-26 | Gilead Sciences, Inc. | Antiviral compounds |
| US11697666B2 (en) | 2021-04-16 | 2023-07-11 | Gilead Sciences, Inc. | Methods of preparing carbanucleosides using amides |
Also Published As
| Publication number | Publication date |
|---|---|
| BR9609013A (en) | 1999-12-14 |
| CZ392797A3 (en) | 1999-04-14 |
| CA2220642A1 (en) | 1996-12-19 |
| EP0832091A1 (en) | 1998-04-01 |
| EP0832091B1 (en) | 2004-01-14 |
| KR19990022740A (en) | 1999-03-25 |
| HUP9802193A2 (en) | 1999-05-28 |
| US5674998A (en) | 1997-10-07 |
| EA199800009A1 (en) | 1998-06-25 |
| OA10639A (en) | 2002-09-17 |
| PL323904A1 (en) | 1998-04-27 |
| AU6178396A (en) | 1996-12-30 |
| ATE257841T1 (en) | 2004-01-15 |
| DE69631330T2 (en) | 2004-10-28 |
| JPH11507387A (en) | 1999-06-29 |
| NO975585D0 (en) | 1997-12-03 |
| AP9701165A0 (en) | 1998-01-31 |
| IS4621A (en) | 1997-11-26 |
| BG102163A (en) | 1998-10-30 |
| TR199701539T1 (en) | 1998-04-21 |
| MX9709859A (en) | 1998-08-30 |
| CN1190401A (en) | 1998-08-12 |
| HUP9802193A3 (en) | 1999-10-28 |
| DE69631330D1 (en) | 2004-02-19 |
| IL122335A0 (en) | 1998-04-05 |
| NO975585L (en) | 1998-02-05 |
| SK166097A3 (en) | 1999-06-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0832091B1 (en) | C-4' modified adenosine kinase inhibitors | |
| US5763596A (en) | C-4' modified adenosine kinase inhibitors | |
| US5763597A (en) | Orally active adenosine kinase inhibitors | |
| US5721356A (en) | Orally active adenosine kinase inhibitors | |
| US5795977A (en) | Water soluble adenosine kinase inhibitors | |
| US5726302A (en) | Water soluble adenosine kinase inhibitors | |
| EP0496617B1 (en) | Adenosine kinase inhibitors | |
| US5646128A (en) | Methods for treating adenosine kinase related conditions | |
| US5506347A (en) | Lyxofuranosyl analogues of adenosine | |
| Cottam et al. | New adenosine kinase inhibitors with oral antiinflammatory activity: synthesis and biological evaluation | |
| US6211158B1 (en) | Desazapurine-nucleotide derivatives, processes for the preparation thereof, pharmaceutical compositions containing them and the use thereof for nucleic acid sequencing and as antiviral agents | |
| US6153594A (en) | 5'-O-acylated antiviral nucleosides | |
| DE3924424A1 (en) | NUCLEOSIDE DERIVATIVES, METHOD FOR THE PRODUCTION THEREOF, THEIR USE AS A MEDICINAL PRODUCT AND THEIR USE IN THE SEQUENCING OF NUCLEIC ACID | |
| AU665184B2 (en) | Adenosine kinase inhibitors | |
| US5864033A (en) | Adenosine kinase inhibitors | |
| MXPA97009656A (en) | Soluble adenosin kinase inhibitors in a | |
| MXPA97009773A (en) | Inhibitors of adenosin cinasa oralmente acti | |
| IE920190A1 (en) | Adenosine kinase inhibitors | |
| MXPA94000878A (en) | Inhibitors of the adenosin kinase, which include derivatives of lixofurans |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 96195383.7 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BB BG BR BY CA CH CN CZ DE DK EE ES FI GB GE HU IL IS JP KE KG KP KR KZ LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK TJ TM TR TT UA UG UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2220642 Country of ref document: CA Ref document number: 2220642 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 310893 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1996919442 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 1997 502290 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 166097 Country of ref document: SK Ref document number: PV1997-3927 Country of ref document: CZ Ref document number: 97/01539 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/1997/009859 Country of ref document: MX Ref document number: 1019970709182 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1199800013 Country of ref document: VN Ref document number: 199800009 Country of ref document: EA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996919442 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019970709182 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1997-3927 Country of ref document: CZ |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV1997-3927 Country of ref document: CZ |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1019970709182 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1996919442 Country of ref document: EP |