WO2001040207A1 - Substituted oxazoles and thiazoles derivatives as hppar alpha activators - Google Patents
Substituted oxazoles and thiazoles derivatives as hppar alpha activators Download PDFInfo
- Publication number
- WO2001040207A1 WO2001040207A1 PCT/EP2000/011995 EP0011995W WO0140207A1 WO 2001040207 A1 WO2001040207 A1 WO 2001040207A1 EP 0011995 W EP0011995 W EP 0011995W WO 0140207 A1 WO0140207 A1 WO 0140207A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- phenoxy
- ylcarbonyl
- amino
- thiazol
- Prior art date
Links
- 0 CC(*C(*)(*1)C2CCC2)C1C(O)=O Chemical compound CC(*C(*)(*1)C2CCC2)C1C(O)=O 0.000 description 2
- HZODWYBXBKXJLB-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1C(N)=S Chemical compound CC(C)(C)c(cc1)ccc1C(N)=S HZODWYBXBKXJLB-UHFFFAOYSA-N 0.000 description 1
- KFLJDSFJBIQZEF-UHFFFAOYSA-N CCOC(C(C)(C)Oc1ccc(CCNC(c2c(C)nc(-c3ccc(C(F)(F)F)cc3)[s]2)=O)cc1)=O Chemical compound CCOC(C(C)(C)Oc1ccc(CCNC(c2c(C)nc(-c3ccc(C(F)(F)F)cc3)[s]2)=O)cc1)=O KFLJDSFJBIQZEF-UHFFFAOYSA-N 0.000 description 1
- UDKIGIDOSXIQBK-UHFFFAOYSA-N CCOC(C(C)(C)Oc1ccc(CN)cc1)=O Chemical compound CCOC(C(C)(C)Oc1ccc(CN)cc1)=O UDKIGIDOSXIQBK-UHFFFAOYSA-N 0.000 description 1
- SYDCCXWAZJHWBY-UHFFFAOYSA-N CCOC(C(C)(C)Oc1ccc(CNC(c2c(C)nc(-c(cc3)ccc3OC(F)(F)F)[s]2)=O)cc1)=O Chemical compound CCOC(C(C)(C)Oc1ccc(CNC(c2c(C)nc(-c(cc3)ccc3OC(F)(F)F)[s]2)=O)cc1)=O SYDCCXWAZJHWBY-UHFFFAOYSA-N 0.000 description 1
- QJPTXUPSYJEXOK-UHFFFAOYSA-N CCOC(C(C)(C)Oc1ccc(CNC(c2c(C)nc(-c3ncc(C(F)(F)F)cc3)[s]2)=O)cc1)=O Chemical compound CCOC(C(C)(C)Oc1ccc(CNC(c2c(C)nc(-c3ncc(C(F)(F)F)cc3)[s]2)=O)cc1)=O QJPTXUPSYJEXOK-UHFFFAOYSA-N 0.000 description 1
- LPIXRQSYBTUXOQ-UHFFFAOYSA-N CCOC(c1c(C)nc(-c2ccc(C(F)(F)F)cc2)[s]1)=O Chemical compound CCOC(c1c(C)nc(-c2ccc(C(F)(F)F)cc2)[s]1)=O LPIXRQSYBTUXOQ-UHFFFAOYSA-N 0.000 description 1
- PJCLTRLEYYGFNR-UHFFFAOYSA-N CCc(cc1)ccc1-c1nc(C)c(C(O)=O)[s]1 Chemical compound CCc(cc1)ccc1-c1nc(C)c(C(O)=O)[s]1 PJCLTRLEYYGFNR-UHFFFAOYSA-N 0.000 description 1
- HGJNTTHIDLWEAC-UHFFFAOYSA-N Cc1cc(CN)ccc1O Chemical compound Cc1cc(CN)ccc1O HGJNTTHIDLWEAC-UHFFFAOYSA-N 0.000 description 1
- AIPANIYQEBQYGC-UHFFFAOYSA-N NC(c(cc1)ccc1Br)=S Chemical compound NC(c(cc1)ccc1Br)=S AIPANIYQEBQYGC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/34—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to certain novel compounds.
- the present invention relates to compounds that activate the alpha subtype of the human peroxisome proliferator activated receptor ("hPPAR alpha").
- the present invention also relates to methods for preparing the compounds and methods for prevention or treatment of PPAR alpha mediated diseases or conditions.
- HMG CoA reductase inhibitors are useful for treating conditions characterized by high LDL-c levels. It has been shown that lowering LDL-c is not sufficient for reducing the risk of cardiovascular disease in some patients, particularly those with normal LDL-c levels. This population pool is identified by the independent risk factor of low HDL-c.
- the increased risk of cardiovascular disease associated with low HDL-c levels has not yet been successfully addressed by drug therapy (i.e., currently there are no drugs on the market that are useful for raising HDL-c >40%).
- Syndrome X (including metabolic syndrome) is loosely defined as a collection of abnormalities including hyperinsulinemia, obesity, elevated levels of trigycerides, uric acid, fibrinogen, small dense LDL-c particles, and plasminogen activator inhibitor 1 (PAI-1), and decreased levels of HDL-c.
- abnormalities including hyperinsulinemia, obesity, elevated levels of trigycerides, uric acid, fibrinogen, small dense LDL-c particles, and plasminogen activator inhibitor 1 (PAI-1), and decreased levels of HDL-c.
- NIDDM is described as insulin resistance which in turn causes anomalous glucose output and a decrease in glucose uptake by skeletal muscle. These factors eventually lead to impaired glucose tolerance (IGT) and hyperinsulinemia.
- ITT impaired glucose tolerance
- Peroxisome Proliferator Activated Receptors are orphan receptors belonging to the steroid/retinoid receptor superfamily of ligand- activated transcription factors. See, for example, Willson, T. M. and Wahli, W., Curr. Opin. Chem. Biol., (1997), Vol. 1, pp 235-241.
- PPAR-alpha Three mammalian Peroxisome Proliferator-Activated Receptors have been isolated and termed PPAR-alpha, PPAR-gamma, and PPAR-delta (also known as NUC1 or PPAR-beta). These PPARs regulate expression of target genes by binding to DNA sequence elements, termed PPAR response elements (PPRE).
- PPRE PPAR response elements
- PPRE's have been identified in the enhancers of a number of genes encoding proteins that regulate lipid metabolism suggesting that PPARs play a pivotal role in the adipogenic signaling cascade and lipid homeostasis (H.
- Fibrates are a class of drugs which may lower serum triglycerides 20- 50%, lower LDL-c 10-15%, shift the LDL particle size from the more atherogenic small dense to normal dense LDL-c, and increase HDL-c 10-15%.
- Experimental evidence indicates that the effects of fibrates on serum lipids are mediated through activation of PPAR alpha. See, for example, B. Staels et al., Curr.
- PPAR alpha activation results in transcription of enzymes that increase fatty acid catabolism and decrease de- novo fatty acid synthesis in the liver resulting in decreased triglyceride synthesis and VLDL-c production/secretion.
- PPAR alpha activation decreases production of apoC-lll.
- Reduction in apoC-lll, an inhibitor of LPL activity increases clearance of VLDL-c.
- PPAR alpha ligands may be useful for the treatment of dyslipidemia and cardiovascular disorders, see Fruchart, J.C., Duriez, P., and Staels, B., Curr. Opin. Lipidol.
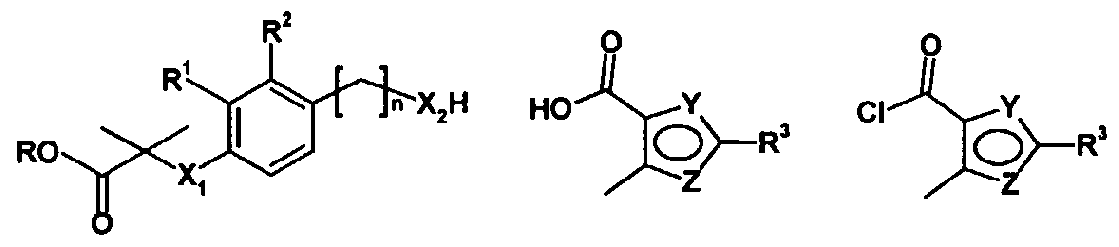
- X represents O or S
- R 1 and R 2 independently represent H, halogen, -CH 3 and -OCH 3 ; n represents 1 or 2;
- X 2 represents NH, NCH 3 or O
- One of Y and Z is N, and the other is O or S;
- R 3 represents phenyl or pyridyl (wherein the N is in position 2 or 3) and is optionally substituted by one or more halogen, NO 2 , NH 2 , CF 3 , OCF 3 , OC ⁇ straight or branched alkyl, C ⁇ straight or branched alkyl, alkenyl or alkynyl with the provision that when R 3 is pyridyl, the N is unsubstituted;
- R 4 represents CF 3 or CH 3
- the present invention discloses a method for prevention or treatment of a human PPAR alpha ("hPPAR alpha”) mediated disease or condition comprising administration of a therapeutically effective amount of a compound of this invention.
- hPPAR alpha mediated diseases or conditions include dyslipidemia including associated diabetic dyslipidemia and mixed dyslipidemia, syndrome X (as defined in this application this embraces metabolic syndrome), heart failure, hypercholesteremia, cardiovascular disease including atherosclerosis, arteriosclerosis, and hypertriglyceridemia, type II diabetes mellitus, type I diabetes, insulin resistance, hyperlipidemia, and regulation of appetite and food intake in subjects suffering from disorders such as obesity, anorexia bulimia, and anorexia nervosa.
- Other diseases or conditions include inflammation.
- the compounds of this invention are useful in the treatment and prevention of cardiovascular diseases and conditions including atherosclerosis, arteriosclerosis, hypertriglyceridemia, and mixed dyslipidaemia.
- the present invention provides pharmaceutical compositions comprising a compound of the invention, preferably in association with a pharmaceutically acceptable diluent or carrier.
- the present invention provides a compound of the invention for use in therapy, and in particular, in human medicine.
- the present invention provides the use of a compound of the invention for the manufacture of a medicament for the treatment of a hPPAR alpha mediated disease or condition.
- the present invention provides a method of treatment of a patient suffering from a hPPAR alpha mediated disease or condition comprising the administration of a therapeutically effective amount of a compound of the invention.
- a compound of the invention means a compound of formula (I) or a pharmaceutically acceptable salt, solvate, or hydrolyzable ester thereof.
- hydrolyzable esters are included in the scope of this invention, the acids are preferred because the data suggests that while the esters are useful compounds, it may actually be the acids to which they hydrolyze that are the active compounds.
- Esters that hydrolyze readily can produce the carboxylic acid in the assay conditions or in vivo.
- the carboxylic acid is active in both the binding and transient transfection assays, while the ester does not usually bind well but is active in the transient transfection assay presumably due to hydrolysis.
- Preferred hydrolysable esters are C ⁇ alkyl esters wherein the alkyl group may be straight chain or branched chain. Methyl or ethyl esters are more preferred.
- X represents O.
- R 1 and R 2 represents H with R 1 and R 2 both representing H being more preferred.
- n 1
- X 2 represents NH
- Z represents N.
- Y represents S.
- R 3 is phenyl, optionally substituted.
- R 3 is mono or disubstituted.
- R 3 preferably is monosubstituted in the para position and is more preferably phenyl.
- a preferred substituent is CF 3 .
- R 4 represents CH 3 .
- preferred compounds of this invention include those in which several or each variable in Formula (I) is selected from the preferred, more preferred, or most preferred groups for each variable. Therefore, this invention is intended to include all combinations of preferred, more preferred, and most preferred groups.
- the compounds of formula (I) are hPPAR alpha agonists.
- agonist or “activating compound”, or “activator”, or the like, is meant those compounds which have a pKi of at least 6.0 to the relevant PPAR, for example hPPAR alpha, in the binding assay described below, and which achieve at least 50% activation of the relevant PPAR relative to the appropriate indicated positive control in the transfection assay described below at concentrations of 10 '5 M or less. More preferably, the compounds of this invention achieve 50% activation of human PPAR alpha in the transfection assay at concentrations of 10 "7 M or less. Most preferably, the compounds of formula (I) are selective hPPAR alpha agonists.
- a "selective hPPAR alpha agonist” is a hPPAR alpha agonist whose EC 50 for PPAR alpha is at least 10 fold lower than its EC 50 for PPAR gamma and PPAR delta. Such selective compounds may be referred to as "10-fold selective.”
- EC 50 is defined in the transfection assay described below and is the concentration at which a compound achieves 50% of its maximum activity. Most preferred compounds are greater than 100-fold selective hPPAR alpha agonists.
- Preferred compounds of the invention include:
- More preferred compounds of the invention include:
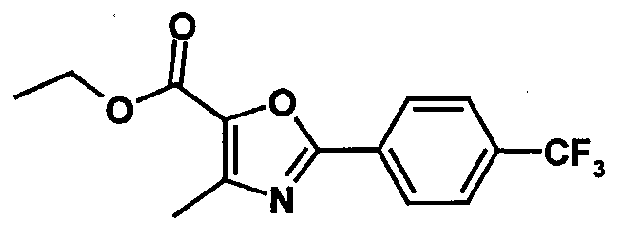
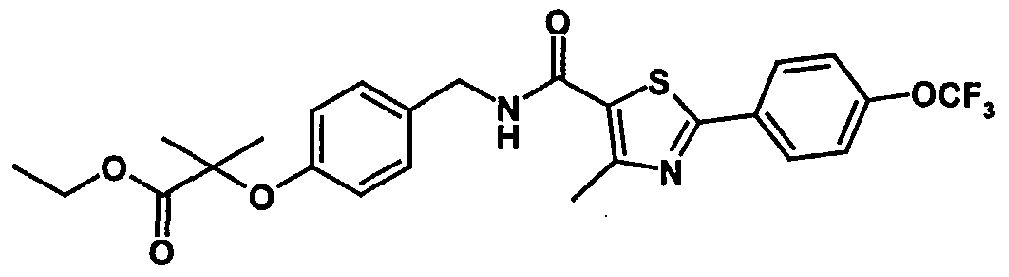
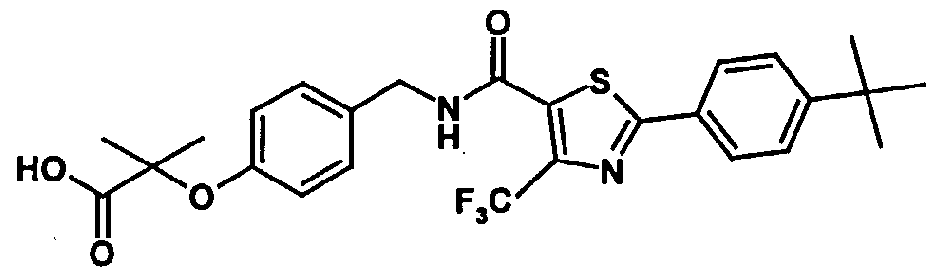
- a particularly preferred compound of the invention is 2-methyl-2-[4- ⁇ [(4- methyl-2-[4-trifluoromethylphenyl]thiazol-5-yl-carbonyl)amino]methyl ⁇ phenoxy] propionic acid.
- the preferred compound listed above is a selective hPPAR alpha agonist.
- the compounds of the present invention may also be utilized in the form of a pharmaceutically acceptable salt or solvate thereof.
- physiologically acceptable salts of the compounds of formula (I) include conventional salts formed from pharmaceutically acceptable inorganic or organic acids or bases as well as quaternary ammonium acid addition salts.
- suitable acid salts include hydrochloric, hydrobromic, sulfuric, phosphoric, nitric, perchloric, fumaric, acetic, propionic, succinic, glycolic, formic, lactic, maleic, tartaric, citric, palmoic, malonic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, fumaric, toluenesulfonic, methanesulfonic, naphthalene-2-sulfonic, benzenesulfonic hydroxynaphthoic, hydroiodic, malic, steroic, tannic and the like.
- acids such as oxalic, while not in themselves pharmaceutically acceptable, may be useful in the preparation of salts useful as intermediates in obtaining the compounds of the invention and their pharmaceutically acceptable salts.
- suitable basic salts include sodium, lithium, potassium, magnesium, aluminium, calcium, zinc, N,N'-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, N-methylglucamine and procaine salts.
- References hereinafter to a compound according to the invention include both compounds of formula (I) and their pharmaceutically acceptable salts and solvates.
- compositions are conveniently administered in the form of pharmaceutical compositions.
- Such compositions may conveniently be presented for use in conventional manner in admixture with one or more physiologically acceptable carriers or excipients.
- compositions of the present invention may be therapeutically administered as the raw chemical, it is preferable to present the active ingredient as a pharmaceutical formulation.
- the carrier(s) must be "acceptable” in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- the present invention further provides for a pharmaceutical formulation comprising a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof together with one or more pharmaceutically acceptable carriers therefore and, optionally, other therapeutic and/or prophylactic ingredients.
- the formulations include those suitable for oral, parenteral (including subcutaneous e.g. by injection or by depot tablet, intradermal, intrathecal, intramuscular e.g. by depot and intravenous), rectal and topical (including dermal, buccal and sublingual) administration although the most suitable route may depend upon for example the condition and disorder of the recipient.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing into association the compounds ("active ingredient") with the carrier which constitutes one or more accessory ingredients. In general the formulations are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers or finely divided solid carriers or both and then, if necessary, shaping the product into the desired formulation.
- Formulations suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets (e.g. chewable tablets in particular for paediatric administration) each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous liquid or a non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion.
- the active ingredient may also be presented as a bolus, electuary or paste.
- a tablet may be made by compression or moulding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a other conventional excipients such as binding agents, (for example, syrup, acacia, gelatin, sorbitol, tragacanth, mucilage of starch or polyvinylpyrrolidone), fillers (for example, lactose, sugar, microcrystalline cellulose, maize-starch, calcium phosphate or sorbitol), lubricants (for example, magnesium stearate, stearic acid, talc, polyethylene glycol or silica), disintegrants (for example, potato starch or sodium starch glycollate) or wetting agents, such as sodium lauryl sulfate.
- binding agents for example, syrup, acacia, gelatin, sorbitol, tragacanth, m
- Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- the tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein.
- the tablets may be coated according to methods well-known in the art.
- the compounds of the present invention may be incorporated into oral liquid preparations such as aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, for example.
- formulations containing these compounds may be presented as a dry product for constitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as suspending agents such as sorbitol syrup, methyl cellulose, glucose/sugar syrup, gelatin, hydroxyethylcellulose, carboxymethyl cellulose, aluminum stearate gel or hydrogenated edible fats; emulsifying agents such as lecithin, sorbitan mono-oleate or acacia; non- aqueous vehicles (which may include edible oils) such as almond oil, fractionated coconut oil, oily esters, propylene glycol or ethyl alcohol; and preservatives such as methyl or propyl p-hydroxybenzoates or sorbic acid.
- Such preparations may also be formulated as suppositories, e.g., containing conventional suppository bases such as cocoa butter or other glycerides.
- Formulations for parenteral administration include aqueous and non- aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the formulations may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilised) condition requiring only the addition of a sterile liquid carrier, for example, water-for-injection, immediately prior to use.
- a sterile liquid carrier for example, water-for-injection
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- Formulations for rectal administration may be presented as a suppository with the usual carriers such as cocoa butter, hard fat or polyethylene glycol.
- Formulations for topical administration in the mouth include lozenges comprising the active ingredient in a flavoured basis such as sucrose and acacia or tragacanth, and pastilles comprising the active ingredient in a basis such as gelatin and glycerin or sucrose and acacia.
- the compounds may also be formulated as depot preparations. Such long acting formulations may be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection.
- the compounds may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- formulations may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavouring agents.
- treatment extends to prophylaxis as well as the treatment of established diseases or symptoms.
- amount of a compound of the invention required for use in treatment will vary with the nature of the condition being treated and the age and the condition of the patient and will be ultimately at the discretion of the attendant physician or veterinarian.
- doses employed for adult human treatment will typically be in the range of 0.02-5000 mg per day, preferably 1-1500 mg per day.
- the desired dose may conveniently be presented in a single dose or as divided doses administered at appropriate intervals, for example as two, three, four or more sub-doses per day.
- the formulations according to the invention may contain between 0.1-99% of the active ingredient, conveniently from 30-95% for tablets and capsules and 3-50% for liquid preparations.
- the compound of formula (I) for use in the instant invention may be used in combination with other therapeutic agents for example, statins and/or other lipid lowering drugs for example MTP inhibitors and LDLR upregulators.
- the compounds of the invention may also be used in combination with antidiabetic agents, e.g. metformin, sulfonylureas and/or PPAR gamma agonists (for example thiazolidinediones such as e.g. Pioglitazone and Rosiglitazone).
- antidiabetic agents e.g. metformin, sulfonylureas and/or PPAR gamma agonists (for example thiazolidinediones such as e.g. Pioglitazone and Rosiglitazone).
- antihypertensive agents such as calcium channel antagonists and ACE inhibitors.
- the invention thus provides in a further aspect the use of a combination comprising a compound of formula (I) with a further therapeutic agent in the treatment of a hPPAR alpha mediated disease.
- a combination comprising a compound of formula (I) with a further therapeutic agent in the treatment of a hPPAR alpha mediated disease.
- the compounds of formula (I) may be administered either sequentially or simultaneously by any convenient route.
- compositions comprising a combination as defined above optimally together with a pharmaceutically acceptable carrier or excipient comprise a further aspect of the invention.
- the individual components of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations.
- the two compounds When combined in the same formulation it will be appreciated that the two compounds must be stable and compatible with each other and the other components of the formulation and may be formulated for administration. When formulated separately they may be provided in any convenient formulation, conveniently in such a manner as are known for such compounds in the art.
- each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art.
- Compounds of this invention may be conveniently prepared by a general process wherein a moiety like (A) is coupled to an acid (B) using a peptide coupling reaction or by alkylation of (A) using a suitable non nucleophilic amine with an acid chloride (C).
- R is 1-6 alkyl which can be hydrolyzed off to give an acid of Formula (I), or if readily hydrolyzable, the resulting ester can be administered.
- Compounds of the invention may be made by an alternative method in which compounds of formula (D) are reacted with ethyl 2-bromo-2 methyl propionate to produce the ethyl ester of the compound of formula (I) which may be hydrolysed to produce the free acid.
- Compounds of formula (D) may be prepared from the reaction between compounds of formula (B) and compounds of formula (E) with HOBT / EDC / NEt 3 when X 2 is NH or NCH 3 or DIC / DMAP / NEt 3 when X 2 is O.
- Example 1 To Example 1 (680mg, 1.39 mmol) in MeOH was added 1N NaOH (1.6 mL, 1.6 mmol) and the reaction stirred at 60°C. After 18h, the reaction cooled to rt and the solvent evaporated. The residue was treated with 1 N HCI, extracted 3 x 20 mL THF and the solvent removed under vacuum. 500mg (75%). The title compound was precipitated as a white solid from a minimum volume of CH 2 CI 2 and pentane. mp: changes form between 60-70°C; LC/MS (m/z): 477.22 (100%, AP-), 479.12 (100%, AP+); anal.
- example 5 To a solution of example 5 (300 mg, 0.6 mmol) in 50 mL of EtOH was added 692 ⁇ L (1.2 equiv.) of NaOH (1N) and the mixture stirred at 60°C for 18h.
- Example 9 was reacted as described in general procedure 8 to afford the title compound as a white solid (74%). MS m/z 493 (M+1)
- Example 12 was reacted as described in general procedure 8 to afford the title compound as a white solid (22%). MS m/z 466 (M+1)
- Example 14 was reacted as described in general procedure 8 to afford the title compound as a white solid (100%). MS m/z 453 (M+1)
- Example 16 was reacted as described in general procedure 8 to afford the title compound as a yellow solid (34%).
- Example 18 was reacted as described in general procedure 8 to afford the title compound as a yellow solid (80%). MS m/z 426 (M+1)
- Example 2 0 was reacted as described in general procedure 8 to afford the title compound as a white solid (84%). MS m/z 480 (M+1)
- Example 22
- Example 22 was reacted as described in general procedure 8 to afford the title compound as a white solid (70%). MS (AP-) m/z 495 (M-1)
- Example 24 was reacted as described in general procedure 8 to afford the title compound as a white solid (90%). MS m/z 489
- Example 26 was reacted as described in general procedure 8 to afford the title compound as a white solid (62%). MS m/z 439 (M+1)
- Example 28 was reacted as described in general procedure 8 to afford the title compound as a white solid (46%). Mp 179°C
- Example 30 was reacted as described in general procedure 8 to afford the title compound as a white solid (72%). Mp 159°C; MS m/z 429 (M+1)
- Example 32 was reacted as described in general procedure 8 to afford the title compound as a white solid (30%).
- Example 34 was reacted as described in general procedure 8 to afford the title compound as a brown viscous oil (45%).
- Example 36 was reacted as described in general procedure 8 to afford the title compound as a beige solid (51%). MS m/z 441 (M+1)
- Example 38 was reacted as described in general procedure 8 to afford the title compound as a pale rose solid (44%). MS (AP-) m/z 433 (M-1) Example 40:
- Example 40 was reacted as described in general procedure 8 to afford the title compound as a clear oil that precipitated in pentane as a white solid (19%). Chromatographed: CH 2 CI 2 /MeOH (95:5), then CH 2 CI 2 /MeOH/AcOH (95:5:2mL).
- Example 42 was reacted as described in general procedure 8 to afford the title compound as a clear oil that precipitated in pentane as a white solid (100%).
- Example 44 was reacted as described in general procedure 8 to afford the title compound as a yellow solid (98%). MS (AP-) m/z 461 (M-1) Example 46:
- Example 46 was reacted as described in general procedure 8 to afford the title compound as a white solid (11%; mixture of the free base and hydrochloride salt).
- Example 48 was reacted as described in general procedure 8 to afford the title compound as a white solid (53%). MS m/z 508
- Example 50 was reacted as described in general procedure 8 to afford the title compound as a white solid (93%). MS m/z 493 (M+1)
- Example 52
- Example 52 was reacted as described in general procedure 8 to afford the title compound as a white solid (85%). MS m/z 467 (M+1)
- Example 54 was reacted as described in general procedure 8 to afford the title compound as a white solid (39%). MS m/z 479 (M+1)
- Example 56 was reacted as described in general procedure 8 to afford the title compound as a white solid (74%). MS m/z 479 (M+1)
- Radioligand 2-(4-(2-(2,3-Ditritio-1 -heptyl-3-(2,4- difluorophenyl)ureido)ethyl) phenoxy)-2-methylbutanoic acid
- a solution of radioligand precursor prepared above (10 mg) in anhydrous DMF (3.5 mL) was transferred to a reaction vessel containing 10 % Pd/C (9.8 mg).
- the reaction vessel was evacuated and degassed via one freeze-thaw- evacuation cycle and then exposed to tritium gas (10.1 Ci). After 4h, the mixture was filtered through celite, evaporated and the residue dissolved in acetonitrile.
- a portion of this solution (0.8 mL, 26.6 mCi) was purified by HPLC (Dynamax C8, 25 min gradient from 4:1 acetonitrile:0.1 %TFA to 9:1 acetonitrile: 0.1 % TFA, 235 nm). Fractions containg pure material were combined and evaporated under nitrogen. The residue was redissolved in acetonitrile to provide a solution of the title compound (82.0 Ci/mmol, radiochemical purity, 99%).
- This compound was used as a positive control for PPAR delta in the transfaction assay and may be prepared as demonstrated below:
- This compound was used as a PPARalpha reference in the transfection assays described below and as prepared according to the following method:
- PPAR ligand binding domain (LBD) was expressed in E. coli as polyHis tagged fusion proteins and purified. The LBD was then labeled with biotin and immobilized on streptavidin-modified scintillation proximity beads.
- the ligand binding domains for murine and human PPAR alpha, PPAR gamma, and PPAR delta were each fused to the yeast transcription factor GAL4 DNA binding domain.
- CV-1 cells were transiently transfected with expression vectors for the respective PPAR chimera along with a reporter construct containing five copies of the GAL4 DNA binding site driving expression of secreted placental alkaline phosphatase (SPAP) and ⁇ -galactosidase. After 16 h, the medium was exchanged to DME medium supplemented with 10% delipidated fetal calf serum and the test compound at the appropriate concentration.
- the positive control in the hPPAR alpha assays was 2-(2-methyl-3-[3- ⁇ 3-(4-cyclohexylamino)-[6-(4-fluorophenylpiperazin-1- yl)][1 ,3,5]triazin-2-ylamino ⁇ propyl]phenylthio)-2-methylpropionic acid.
- the positive control for PPAR delta assays was 2- ⁇ 2-methyl-4-[( ⁇ 4-methyl-2- ⁇ trifluoromethyl)phenyl]-1 ,3-thiazol-5-yl ⁇ methyl)sulfanyl]phenoxy ⁇ acetic acid.
Abstract
Description























































































































Claims

Priority Applications (17)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/148,765 US6518290B1 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazoles derivatives as HPPAr alpha activators |
| BR0016067-9A BR0016067A (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazole derivatives as activators of hppar alfa |
| DK00983189T DK1244642T3 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazole and thiazole derivatives as HPPAR alpha activators |
| IL14958200A IL149582A0 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazoles derivative as hppar alpha activators |
| DE60010333T DE60010333T2 (en) | 1999-12-02 | 2000-11-30 | SUBSTITUTED OXAZOLE AND THIAZOLE DERIVATIVES AS HPPAR-ALPHA ACTIVATORE |
| CA002393190A CA2393190A1 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazoles derivatives as hppar alpha activators |
| MXPA02005456A MXPA02005456A (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazoles derivatives as hppar alpha activators. |
| AT00983189T ATE265442T1 (en) | 1999-12-02 | 2000-11-30 | SUBSTITUTED OXAZOLE AND THIAZOLE DERIVATIVES AS HPPAR-ALPHA ACTIVATORS |
| PL00356772A PL356772A1 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazoles derivatives as hppar alpha activators |
| JP2001541891A JP3884290B2 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazole and thiazole derivatives as HPPAR alpha activators |
| AU20030/01A AU758758B2 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazoles derivatives as HPPAR alpha activators |
| HU0203532A HUP0203532A3 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazoles derivatives as hppar alpha activators, pharmaceutical compositions containing them and their use |
| EP00983189A EP1244642B1 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazoles derivatives as hppar alpha activators |
| SI200030421T SI1244642T1 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazoles derivatives as hppar alpha activators |
| IL149582A IL149582A (en) | 1999-12-02 | 2002-05-09 | SUBSTITUTED OXAZOLE AND THIAZOLE DERIVATIVES, PHARMACEUTICAL COMPOSITIONS COMPRISING THE SAME AND THEIR USE FOR THE MANUFACTURE OF MEDICAMENTS FOR THE TREATMENT OF hPPAR ALPHA DISEASE OR CONDITION |
| NO20022467A NO323135B1 (en) | 1999-12-02 | 2002-05-24 | Substituted oxazole and thiazole derivatives as hPPAR alpha activators, a pharmaceutical composition comprising these and their use in the manufacture of a medicament |
| HK02109084.6A HK1047435B (en) | 1999-12-02 | 2002-12-14 | Substituted oxazoles and thiazoles derivatives as hppar alpha activators |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9928561.1 | 1999-12-02 | ||
| GBGB9928561.1A GB9928561D0 (en) | 1999-12-02 | 1999-12-02 | Chemical compounds |
| GB0003500A GB0003500D0 (en) | 2000-02-15 | 2000-02-15 | Chemical compounds |
| GB0003500.6 | 2000-02-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001040207A1 true WO2001040207A1 (en) | 2001-06-07 |
Family
ID=26243654
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2000/011995 WO2001040207A1 (en) | 1999-12-02 | 2000-11-30 | Substituted oxazoles and thiazoles derivatives as hppar alpha activators |
Country Status (27)
| Country | Link |
|---|---|
| US (1) | US6518290B1 (en) |
| EP (1) | EP1244642B1 (en) |
| JP (1) | JP3884290B2 (en) |
| KR (1) | KR100706735B1 (en) |
| CN (1) | CN1184215C (en) |
| AR (1) | AR035631A1 (en) |
| AT (1) | ATE265442T1 (en) |
| AU (1) | AU758758B2 (en) |
| BR (1) | BR0016067A (en) |
| CA (1) | CA2393190A1 (en) |
| CO (1) | CO5251452A1 (en) |
| CZ (1) | CZ20021903A3 (en) |
| DE (1) | DE60010333T2 (en) |
| DK (1) | DK1244642T3 (en) |
| ES (1) | ES2219423T3 (en) |
| GC (1) | GC0000215A (en) |
| HK (1) | HK1047435B (en) |
| HU (1) | HUP0203532A3 (en) |
| IL (2) | IL149582A0 (en) |
| MX (1) | MXPA02005456A (en) |
| NO (1) | NO323135B1 (en) |
| PE (1) | PE20011010A1 (en) |
| PL (1) | PL356772A1 (en) |
| PT (1) | PT1244642E (en) |
| TR (2) | TR200201473T2 (en) |
| TW (1) | TW555753B (en) |
| WO (1) | WO2001040207A1 (en) |
Cited By (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002009682A2 (en) * | 2000-08-01 | 2002-02-07 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Use of fibrates for the preparation of a medicament useful in the treatment of congestive heart failure |
| WO2002014291A1 (en) | 2000-08-11 | 2002-02-21 | Nippon Chemiphar Co.,Ltd. | PPARδ ACTIVATORS |
| WO2002038553A2 (en) * | 2000-11-10 | 2002-05-16 | Eli Lilly And Company | Triazole derivatives and their use as peroxisome proliferator activated receptor alpha agonists |
| WO2002050047A1 (en) * | 2000-12-20 | 2002-06-27 | Glaxo Group Limited | Substitued oxazoles and thiazoles as hppar alpha agonists |
| WO2002059098A1 (en) * | 2000-12-20 | 2002-08-01 | Glaxo Group Limited | Thiazole and oxazole derivatives as activators of human peroxisome proliferator activated receptors |
| WO2002062774A1 (en) * | 2000-12-20 | 2002-08-15 | Glaxo Group Limited | Thiazole derivatives for treating ppar related disorders |
| WO2002096893A1 (en) * | 2001-05-31 | 2002-12-05 | Glaxo Group Limited | Process for preparing a thialzole ppar-ligand and polymorphs thereof |
| WO2002096894A1 (en) * | 2001-05-31 | 2002-12-05 | Glaxo Group Limited | Thiazole or oxazole derivatives which are useful in the treatment of cardiovascular and related diseases |
| WO2003016291A1 (en) * | 2001-08-10 | 2003-02-27 | Nippon Chemiphar Co., Ltd. | ACTIVATOR FOR PEROXISOME PROLIFERATOR-RESPONSIVE RECEPTOR δ |
| WO2003037332A1 (en) * | 2001-10-12 | 2003-05-08 | Bayer Pharmaceuticals Corporation | Phenyl substituted 5-membered nitrogen containing heterocycles for the treatment of obesity |
| WO2003040107A1 (en) * | 2001-09-24 | 2003-05-15 | Bayer Pharmaceuticals Corporation | Imidazole-4-carboxamide derivatives, preparation and use thereof for treatment of obesity |
| WO2003074495A1 (en) * | 2002-03-01 | 2003-09-12 | Smithkline Beecham Corporation | Hppars activators |
| WO2004000785A2 (en) * | 2002-06-19 | 2003-12-31 | Smithkline Beecham Corporation | Phenyloxyalkanonic acid derivatives as hppar activators |
| WO2004103997A1 (en) * | 2003-05-21 | 2004-12-02 | Pfizer Products Inc. | TETRAHYDROISOQUINOLINE DERIVATIVES AS PPAR-α ACTIVATORS |
| KR100474202B1 (en) * | 2002-05-04 | 2005-03-08 | 강헌중 | Process for preparing thiazol derivative and the intermediate compounds for preparing the same |
| WO2005012273A3 (en) * | 2003-07-30 | 2005-05-12 | Abbott Lab | Process for the preparation of substituted thiazoles |
| WO2005049578A1 (en) * | 2003-11-17 | 2005-06-02 | Smithkline Beecham Corporation | Substituted pyrazoles as ppar agonists |
| US6933308B2 (en) | 2002-12-20 | 2005-08-23 | Bristol-Myers Squibb Company | Aminoalkyl thiazole derivatives as KCNQ modulators |
| WO2006013939A1 (en) * | 2004-08-05 | 2006-02-09 | Daiichi Pharmaceutical Co., Ltd. | Pyrazole derivatives |
| US7105551B2 (en) | 2000-12-20 | 2006-09-12 | Smithkline Beecham Corporation | Thiazole derivatives for treating PPAR related disorders |
| JP2006520755A (en) * | 2003-02-14 | 2006-09-14 | イーライ リリー アンド カンパニー | Sulfonamide derivatives as PPAR modulators |
| US7157479B2 (en) * | 2001-05-31 | 2007-01-02 | Glaxo Group Limited | Oxazol/thiazol-derivatives activators of the hPPAR-alpha receptor |
| WO2007028424A1 (en) * | 2005-02-15 | 2007-03-15 | F. Hoffmann-La Roche Ag | Amide derivatives as ppar activators |
| US7220880B2 (en) | 2002-06-19 | 2007-05-22 | Eli Lilly And Company | Amide linker peroxisome proliferator activated receptor modulators |
| US7229998B2 (en) | 2000-12-20 | 2007-06-12 | Smithkline Beecham Corporation | Thiazole and oxazole derivatives as activators of human peroxisome proliferator activated receptors |
| US7273866B2 (en) | 2002-12-20 | 2007-09-25 | Bristol-Myers Squibb Company | 2-aryl thiazole derivatives as KCNQ modulators |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US7541475B2 (en) | 2003-07-30 | 2009-06-02 | Abbott Laboratories | Substituted thiazoles |
| DE102007063671A1 (en) | 2007-11-13 | 2009-06-25 | Sanofi-Aventis Deutschland Gmbh | New crystalline diphenylazetidinone hydrates, medicaments containing these compounds and their use |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2010071813A1 (en) * | 2008-12-19 | 2010-06-24 | Aryx Therapeutics, Inc. | AGONISTS OF PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR-α |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8404726B2 (en) | 2006-04-18 | 2013-03-26 | Nippon Chemiphar Co. Ltd. | Activating agent for peroxisome proliferator activated receptor δ |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8648208B2 (en) | 2008-04-15 | 2014-02-11 | Nippon Chemiphar Co. Ltd. | Activating agent for peroxisome proliferator activated receptor |
| US8785681B2 (en) | 2007-03-08 | 2014-07-22 | Albireo Ab | 2-substituted-3-phenylpropionic acid derivatives and their use in the treatment of inflammatory bowel disease |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0031109D0 (en) * | 2000-12-20 | 2001-01-31 | Glaxo Group Ltd | Chemical compounds |
| GB0314370D0 (en) * | 2003-06-19 | 2003-07-23 | Glaxo Group Ltd | Chemical compounds |
| DE10331496A1 (en) * | 2003-07-01 | 2005-01-27 | Bayer Cropscience Ag | Process for preparing alkyl difluoroacetoacetates |
| JP2007230868A (en) * | 2004-03-30 | 2007-09-13 | Dai Ichi Seiyaku Co Ltd | Bicyclic compound and medicine using the same |
| US20070208021A1 (en) * | 2004-03-30 | 2007-09-06 | Daiichi Pharmaceutical Co., Ltd. | Phenoxyacetic Acid Derivatives and Drug Comprising The Same |
| DE102004016845A1 (en) * | 2004-04-07 | 2005-10-27 | Bayer Healthcare Ag | Phenylthioacetic acid derivatives and their use |
| EP1734930A2 (en) * | 2004-04-09 | 2006-12-27 | Smithkline Beecham Corporation | Low dose pharmaceutical products |
| MX2007001634A (en) * | 2004-08-11 | 2007-04-23 | Kyorin Seiyaku Kk | Novel cyclic aminobenzoic acid derivative. |
| EP1862464A4 (en) * | 2005-03-23 | 2010-08-25 | Kyorin Seiyaku Kk | Novel cyclic aminophenylalkanoic acid derivative |
| JP3795515B1 (en) | 2005-08-10 | 2006-07-12 | 善典 中川 | Manufacturing method of semiconductor photoelectrochemical cell |
| WO2008035359A2 (en) * | 2006-06-12 | 2008-03-27 | Cadila Healthcare Limited | Oximinophenoxyalkanoic acid and phenylalkanoic acid derivatives |
| KR20150118197A (en) * | 2007-07-19 | 2015-10-21 | 하. 룬트벡 아크티에 셀스카브 | 5-membered heterocyclic amides and related compounds |
| KR100882261B1 (en) | 2007-07-25 | 2009-02-06 | 삼성전기주식회사 | Manufacturing method and system of printed circuit board |
| US8247436B2 (en) | 2010-03-19 | 2012-08-21 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| CN109810071B (en) * | 2019-03-28 | 2023-04-21 | 中国科学院成都生物研究所 | miRNA biosynthesis inhibitor |
| CN110803996B (en) * | 2019-11-13 | 2022-01-11 | 台州市创源工业技术有限公司 | Synthesis method of hydroxybenzylamine |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997036579A1 (en) * | 1996-03-30 | 1997-10-09 | Glaxo Group Limited | Use of agonists of the peroxisome proliferator activated receptor alpha for treating obesity |
| WO1999018066A1 (en) * | 1997-10-02 | 1999-04-15 | Sankyo Company, Limited | Amidocarboxylic acid derivatives |
| WO1999046232A1 (en) * | 1998-03-10 | 1999-09-16 | Ono Pharmaceutical Co., Ltd. | Carboxylic acid derivatives and drugs containing the same as the active ingredient |
-
2000
- 2000-11-28 PE PE2000001264A patent/PE20011010A1/en not_active Application Discontinuation
- 2000-11-29 TW TW089125344A patent/TW555753B/en active
- 2000-11-30 TR TR2002/01473T patent/TR200201473T2/en unknown
- 2000-11-30 PL PL00356772A patent/PL356772A1/en unknown
- 2000-11-30 CN CNB008166315A patent/CN1184215C/en not_active Expired - Fee Related
- 2000-11-30 AT AT00983189T patent/ATE265442T1/en not_active IP Right Cessation
- 2000-11-30 ES ES00983189T patent/ES2219423T3/en not_active Expired - Lifetime
- 2000-11-30 IL IL14958200A patent/IL149582A0/en active IP Right Grant
- 2000-11-30 BR BR0016067-9A patent/BR0016067A/en not_active Application Discontinuation
- 2000-11-30 CZ CZ20021903A patent/CZ20021903A3/en unknown
- 2000-11-30 JP JP2001541891A patent/JP3884290B2/en not_active Expired - Fee Related
- 2000-11-30 AR ARP000106321A patent/AR035631A1/en unknown
- 2000-11-30 KR KR1020027007081A patent/KR100706735B1/en not_active IP Right Cessation
- 2000-11-30 AU AU20030/01A patent/AU758758B2/en not_active Ceased
- 2000-11-30 TR TR2004/01772T patent/TR200401772T4/en unknown
- 2000-11-30 CO CO00091770A patent/CO5251452A1/en not_active Application Discontinuation
- 2000-11-30 WO PCT/EP2000/011995 patent/WO2001040207A1/en active IP Right Grant
- 2000-11-30 EP EP00983189A patent/EP1244642B1/en not_active Expired - Lifetime
- 2000-11-30 HU HU0203532A patent/HUP0203532A3/en unknown
- 2000-11-30 MX MXPA02005456A patent/MXPA02005456A/en active IP Right Grant
- 2000-11-30 DK DK00983189T patent/DK1244642T3/en active
- 2000-11-30 US US10/148,765 patent/US6518290B1/en not_active Expired - Fee Related
- 2000-11-30 DE DE60010333T patent/DE60010333T2/en not_active Expired - Lifetime
- 2000-11-30 PT PT00983189T patent/PT1244642E/en unknown
- 2000-11-30 CA CA002393190A patent/CA2393190A1/en not_active Abandoned
- 2000-12-02 GC GCP20001073 patent/GC0000215A/en active
-
2002
- 2002-05-09 IL IL149582A patent/IL149582A/en not_active IP Right Cessation
- 2002-05-24 NO NO20022467A patent/NO323135B1/en not_active IP Right Cessation
- 2002-12-14 HK HK02109084.6A patent/HK1047435B/en not_active IP Right Cessation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997036579A1 (en) * | 1996-03-30 | 1997-10-09 | Glaxo Group Limited | Use of agonists of the peroxisome proliferator activated receptor alpha for treating obesity |
| WO1999018066A1 (en) * | 1997-10-02 | 1999-04-15 | Sankyo Company, Limited | Amidocarboxylic acid derivatives |
| EP1026149A1 (en) * | 1997-10-02 | 2000-08-09 | Sankyo Company Limited | Amidocarboxylic acid derivatives |
| WO1999046232A1 (en) * | 1998-03-10 | 1999-09-16 | Ono Pharmaceutical Co., Ltd. | Carboxylic acid derivatives and drugs containing the same as the active ingredient |
| EP1067109A1 (en) * | 1998-03-10 | 2001-01-10 | Ono Pharmaceutical Co., Ltd. | Carboxylic acid derivatives and drugs containing the same as the active ingredient |
Cited By (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002009682A2 (en) * | 2000-08-01 | 2002-02-07 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Use of fibrates for the preparation of a medicament useful in the treatment of congestive heart failure |
| WO2002009682A3 (en) * | 2000-08-01 | 2002-04-18 | Sigma Tau Ind Farmaceuti | Use of fibrates for the preparation of a medicament useful in the treatment of congestive heart failure |
| WO2002014291A1 (en) | 2000-08-11 | 2002-02-21 | Nippon Chemiphar Co.,Ltd. | PPARδ ACTIVATORS |
| EP1310494A4 (en) * | 2000-08-11 | 2004-10-20 | Nippon Chemiphar Co | PPAR (delta) ACTIVATORS |
| EP1310494A1 (en) * | 2000-08-11 | 2003-05-14 | Nippon Chemiphar Co., Ltd. | PPAR (delta) ACTIVATORS |
| US7304062B2 (en) | 2000-11-10 | 2007-12-04 | Eli Lilly And Company | Peroxisome proliferator activated receptor alpha agonists |
| WO2002038553A3 (en) * | 2000-11-10 | 2003-05-01 | Lilly Co Eli | Triazole derivatives and their use as peroxisome proliferator activated receptor alpha agonists |
| WO2002038553A2 (en) * | 2000-11-10 | 2002-05-16 | Eli Lilly And Company | Triazole derivatives and their use as peroxisome proliferator activated receptor alpha agonists |
| WO2002062774A1 (en) * | 2000-12-20 | 2002-08-15 | Glaxo Group Limited | Thiazole derivatives for treating ppar related disorders |
| US7229998B2 (en) | 2000-12-20 | 2007-06-12 | Smithkline Beecham Corporation | Thiazole and oxazole derivatives as activators of human peroxisome proliferator activated receptors |
| WO2002059098A1 (en) * | 2000-12-20 | 2002-08-01 | Glaxo Group Limited | Thiazole and oxazole derivatives as activators of human peroxisome proliferator activated receptors |
| US7105551B2 (en) | 2000-12-20 | 2006-09-12 | Smithkline Beecham Corporation | Thiazole derivatives for treating PPAR related disorders |
| US7091225B2 (en) | 2000-12-20 | 2006-08-15 | Smithkline Beecham Corporation | Substituted oxazoles and thiazoles as hPPAR alpha agonists |
| WO2002050047A1 (en) * | 2000-12-20 | 2002-06-27 | Glaxo Group Limited | Substitued oxazoles and thiazoles as hppar alpha agonists |
| US7439259B2 (en) * | 2000-12-20 | 2008-10-21 | Smithkline Beecham Corporation | Thiazole derivatives for treating PPAR related disorders |
| US7449468B2 (en) | 2000-12-20 | 2008-11-11 | Smithkline Beecham Corporation | Thiazole and oxazole derivatives as activators of human peroxisome proliferator activated receptors |
| AU2002246713B2 (en) * | 2000-12-20 | 2004-09-09 | Glaxo Group Limited | Thiazole derivatives for treating PPAR related disorders |
| WO2002096894A1 (en) * | 2001-05-31 | 2002-12-05 | Glaxo Group Limited | Thiazole or oxazole derivatives which are useful in the treatment of cardiovascular and related diseases |
| US7244849B2 (en) | 2001-05-31 | 2007-07-17 | Glaxo Group Limited | Process for preparing a thiazole PPAR-ligand and polymorphs thereof |
| US7157479B2 (en) * | 2001-05-31 | 2007-01-02 | Glaxo Group Limited | Oxazol/thiazol-derivatives activators of the hPPAR-alpha receptor |
| WO2002096893A1 (en) * | 2001-05-31 | 2002-12-05 | Glaxo Group Limited | Process for preparing a thialzole ppar-ligand and polymorphs thereof |
| US6867225B2 (en) | 2001-05-31 | 2005-03-15 | Smithkline Beecham Corporation | Thiazole or oxazole derivatives which are useful in the treatment of cardiovascular and related diseases |
| JP2009102442A (en) * | 2001-08-10 | 2009-05-14 | Nippon Chemiphar Co Ltd | ACTIVATOR OF PEROXISOME PROLIFERATOR-RESPONSIVE RECEPTOR delta |
| KR100901683B1 (en) * | 2001-08-10 | 2009-06-08 | 닛뽕 케미파 가부시키가이샤 | Activator of peroxisome proliferator-activated receptor ? |
| JP2009280622A (en) * | 2001-08-10 | 2009-12-03 | Nippon Chemiphar Co Ltd | ACTIVATOR OF PEROXISOME PROLIFERATOR-RESPONSIVE RECEPTOR delta |
| US7652045B2 (en) | 2001-08-10 | 2010-01-26 | Nippon Chemiphar, Co., Ltd. | Activator of peroxisome proliferator-activated receptor δ |
| WO2003016291A1 (en) * | 2001-08-10 | 2003-02-27 | Nippon Chemiphar Co., Ltd. | ACTIVATOR FOR PEROXISOME PROLIFERATOR-RESPONSIVE RECEPTOR δ |
| US7265137B2 (en) | 2001-08-10 | 2007-09-04 | Nippon Chemiphar Co., Ltd. | Activator of peroxisome proliferator-activated re-ceptor δ |
| US7648999B2 (en) | 2001-08-10 | 2010-01-19 | Nippon Chemiphar Co., Ltd. | Activator for peroxisome proliferator-activated receptor δ |
| US6960601B2 (en) | 2001-09-24 | 2005-11-01 | Bayer Pharmaceuticals Corporation | Preparation and use of imidazole derivatives for treatment of obesity |
| WO2003040107A1 (en) * | 2001-09-24 | 2003-05-15 | Bayer Pharmaceuticals Corporation | Imidazole-4-carboxamide derivatives, preparation and use thereof for treatment of obesity |
| WO2003037332A1 (en) * | 2001-10-12 | 2003-05-08 | Bayer Pharmaceuticals Corporation | Phenyl substituted 5-membered nitrogen containing heterocycles for the treatment of obesity |
| WO2003074495A1 (en) * | 2002-03-01 | 2003-09-12 | Smithkline Beecham Corporation | Hppars activators |
| US7319104B2 (en) | 2002-03-01 | 2008-01-15 | Smithkline Beecham Corporation | hPPARs activators |
| KR100474202B1 (en) * | 2002-05-04 | 2005-03-08 | 강헌중 | Process for preparing thiazol derivative and the intermediate compounds for preparing the same |
| WO2004000785A3 (en) * | 2002-06-19 | 2004-10-14 | Smithkline Beecham Corp | Phenyloxyalkanonic acid derivatives as hppar activators |
| US7220880B2 (en) | 2002-06-19 | 2007-05-22 | Eli Lilly And Company | Amide linker peroxisome proliferator activated receptor modulators |
| US7241793B2 (en) | 2002-06-19 | 2007-07-10 | Smithkline Beecham Corporation | Phenyloxyalkanonic acid derivatives as hPPAR activators |
| WO2004000785A2 (en) * | 2002-06-19 | 2003-12-31 | Smithkline Beecham Corporation | Phenyloxyalkanonic acid derivatives as hppar activators |
| US6933308B2 (en) | 2002-12-20 | 2005-08-23 | Bristol-Myers Squibb Company | Aminoalkyl thiazole derivatives as KCNQ modulators |
| US7273866B2 (en) | 2002-12-20 | 2007-09-25 | Bristol-Myers Squibb Company | 2-aryl thiazole derivatives as KCNQ modulators |
| JP2006520755A (en) * | 2003-02-14 | 2006-09-14 | イーライ リリー アンド カンパニー | Sulfonamide derivatives as PPAR modulators |
| WO2004103997A1 (en) * | 2003-05-21 | 2004-12-02 | Pfizer Products Inc. | TETRAHYDROISOQUINOLINE DERIVATIVES AS PPAR-α ACTIVATORS |
| US6987118B2 (en) | 2003-05-21 | 2006-01-17 | Pfizer Inc. | Tetrahydroisoquinoline derivatives as PPAR-alpha activators |
| US7541475B2 (en) | 2003-07-30 | 2009-06-02 | Abbott Laboratories | Substituted thiazoles |
| WO2005012273A3 (en) * | 2003-07-30 | 2005-05-12 | Abbott Lab | Process for the preparation of substituted thiazoles |
| WO2005049578A1 (en) * | 2003-11-17 | 2005-06-02 | Smithkline Beecham Corporation | Substituted pyrazoles as ppar agonists |
| WO2006013939A1 (en) * | 2004-08-05 | 2006-02-09 | Daiichi Pharmaceutical Co., Ltd. | Pyrazole derivatives |
| WO2007028424A1 (en) * | 2005-02-15 | 2007-03-15 | F. Hoffmann-La Roche Ag | Amide derivatives as ppar activators |
| US8404726B2 (en) | 2006-04-18 | 2013-03-26 | Nippon Chemiphar Co. Ltd. | Activating agent for peroxisome proliferator activated receptor δ |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| US9181181B2 (en) | 2007-03-08 | 2015-11-10 | Albireo Ab | 2-substituted-3-phenylpropionic acid derivatives and their use in the treatment of inflammatory bowel disease |
| US8785681B2 (en) | 2007-03-08 | 2014-07-22 | Albireo Ab | 2-substituted-3-phenylpropionic acid derivatives and their use in the treatment of inflammatory bowel disease |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| DE102007063671A1 (en) | 2007-11-13 | 2009-06-25 | Sanofi-Aventis Deutschland Gmbh | New crystalline diphenylazetidinone hydrates, medicaments containing these compounds and their use |
| US8648208B2 (en) | 2008-04-15 | 2014-02-11 | Nippon Chemiphar Co. Ltd. | Activating agent for peroxisome proliferator activated receptor |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2010071813A1 (en) * | 2008-12-19 | 2010-06-24 | Aryx Therapeutics, Inc. | AGONISTS OF PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR-α |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1244642B1 (en) | Substituted oxazoles and thiazoles derivatives as hppar alpha activators | |
| EP1392665B1 (en) | Thiazole or oxazole derivatives which are useful in the treatment of cardiovascular and related diseases | |
| AU2002312954A1 (en) | Thiazole or oxazole derivatives which are useful in the treatment of cardiovascular and related diseases | |
| US20040147571A1 (en) | Substituted oxazoles and thiazoles as hppar alpha agonists | |
| EP1399430B1 (en) | Oxazol/ thiazol-derivatives activators of the hppar-alpha receptor | |
| AU2002317765A1 (en) | Oxazol/ thiazol-derivatives activators of the hPPAR-alpha receptor | |
| US20070167628A1 (en) | Thiazole-2-carboxamide derivatives for use as hippar agonists in the treatment of i.a. dyslipidemia |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 518794 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 149582 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20030/01 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000983189 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002/04241 Country of ref document: ZA Ref document number: 200204241 Country of ref document: ZA |
|
| ENP | Entry into the national phase |
Ref document number: 2001 541891 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/005456 Country of ref document: MX Ref document number: PV2002-1903 Country of ref document: CZ Ref document number: 2393190 Country of ref document: CA Ref document number: 10148765 Country of ref document: US Ref document number: 2002/01473 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027007081 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 008166315 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027007081 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000983189 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2002-1903 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 20030/01 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 518794 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 518794 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2000983189 Country of ref document: EP |