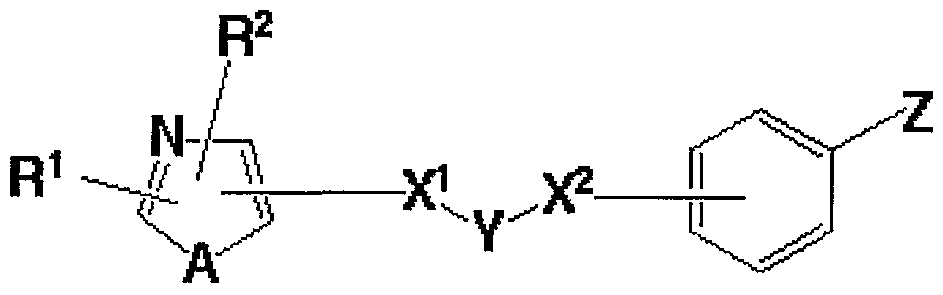明 細 書 ペルォキシゾーム増殖剤応答性受容体 Sの活性化剤 技術分野
本発明はペルォキシゾーム増殖剤応答性受容体 (5の活性化剤に関する。 背景技術
ペルォキシソーム (p e r ox i s ome) は動植物の細胞中に見られる小器 官で、 そのマトリックスには力夕ラーゼをはじめとした種々の酵素が含まれてい る。 ぺゾレオキシソ一ム ί曽殖斉 (p e r ox i s ome p r o l i f e r a t e) r ) は、 このペルォキシゾームの増殖を誘発する物質で抗脂血薬 (フイブラート 類) 、 除草剤、 フ夕ル酸塩可塑剤等の多様な化合物群が知られている。
ィヅセマン ( I s s eman) らによりこのペルォキシゾーム増殖剤によって 活性化される核内受容体が同定され、 ペルォキシソーム増殖剤応答性受容体 (p e r o x i s ome p r o l i f e r a t o r a c t i va t e d r e c e p t o r : PPAR) と命名された。 (Na t u r e, 347 , p 645 - 6 50, 19 90 )
PPAI まこれまで PPARひ、 P P ARァ及び P P A R 5の 3種のサブ · 夕 イブの存在が確認されている。 (P r o c. Na t l . Ac ad. S c i . US A, 9 1, p 733 5 - 73 59 , Γ994) '
上述したフィブラ一ト系薬剤はこのうち P PARひに対しリガンド効果を有し 、 臨床では強い血清 T G (トリグリセリ ド) の低下作用が認められている。
また糖尿病治療薬であるチアゾリジンジオン系化合物 (T r o g l i t a z o n e , R o s i g l i t a z o ne, P i o g l i t a z o ne) は、 PPAR ァのリガンドとして知られている。
P PAR 5活性化作用 (活性化能) を有する薬物としては、 例えば次式、
で表される GW— 2433 (G l ax o We l l c ome) 、 次式、
で表される L— 1 6 504 1 (Me r c k) 或いは次式、
で表される YM— 16638 (山之内製薬) 等が知られている。
GW- 2433はァテローム硬化症の予防及び治療薬としての使用が WO 92 0468に記載され、 L一 165041は糖尿病治療剤ゃ抗肥満薬としての 使用が W〇 97/28 1 15に記載され、 そして YM— 16638については W 099/0481 5に血清コレステロール低下作用、 LD L—コレステロール低 下作用を有する旨の記載がなされている。 更に最近、 P P A R (5のリガンドは抗ガン剤ゃ抗炎症剤としての応用を促す報 告 (JBC, 272 (6) 、 p 3406-3410, 1997 ; C e 11, 99 , p 335— 345 , 1999 ) がなされている。 一方、 本発明化合物である後記一般式 (I I) で表されるォキサゾール誘導体 と類似した構造を有する化合物として次式で表される化合物 A、
が欧州特許 558062号公報に記載されており、 又文献 I (J. Immuno 1. Met hod s, 207 (1) , 23— 31, 1997 ) には次式で表され
る化合物 B、 H
が記載されている。 上記化合物 A及び B、 並びに本発明化合物の後記一般式 (I I) で表されるォ キサゾール誘導体は共にフエノキシ酢酸タイプの化合物であるが、 上記化合物 A 及び Bの酢酸の α位には置換基がなく、 一方、 本発明化合物は α, ひージアルキ ルフエノキシ酢酸タイプの化合物であり、 明確な構造上の相違を有する。
尚、 上記欧州特許 558062号公報には化合物 Αが抗トロンビン剤、 血圧降 下剤等として有用である旨の記載があるが、 この化合物が P PAR 3のリガンド として有用である旨の具体的な記載はない。
また、 上記文献 Iは上記化合物 Bの血糖降下作用に係わるものであるが、 この 化合物が P P A R dのリガンドとして有用である旨の具体的な記載はない。 さらに、 最近 P PAR のァゴニスト作用を有する置換ォキサ (チア) ゾール 誘導体に関する WO 0 1/40207が国際公開され、 また PP AR調節剤とし て使用可能なビアリール基で置換されたォキサ (チア) ゾール誘導体に関する W 001/ 16 120が国際公開されている。
このうち、 WO 01/40207は本願化合物における X1が C ( = 0) NH で、 X2が結合手に相当し、 WO 0 1/16 120は本願化合物における X1が 結合手で、 X2が 0、 S等に相当することから何れも本願化合物とは構造上、 相 違" 5' 。 本発明の目的はペルォキシゾーム増殖剤応答性受容体 (5の活性化作用 (活性化
能) を有する下記一般式 (I) で表される化合物、 下記一般式 (τ ι) で表され るォキサゾール誘導体、 又は下記一般式 (III) で表されるチア、 -ル誘導体を 提供することにある。 発明の開示
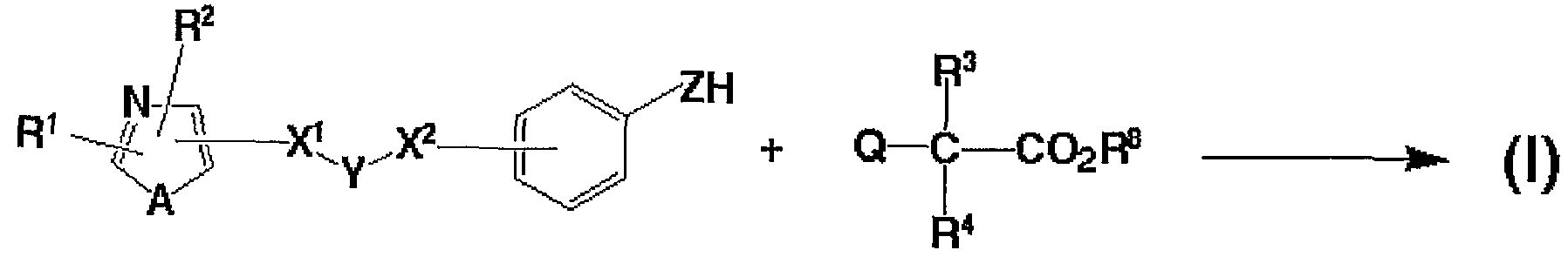
即ち、 本発明は、 次の一般式 (I) 、
(式中、 R1及び R2はそれぞれ独立して水素原子、 炭素数 1〜 8のアルキル基 、 ハロゲン原子で置換された炭素数 1〜 8のアルキル基、 炭素数 2〜 8のァルケ ニル基、 炭素数 2〜 8のアルキニル基、 3〜7員環のシクロアルキル基、 3〜7 員環のシクロアルキル基で置換された炭素数 1〜 8のアルキル基、 置換基を有し ていても良いァリ一ルアルキル基 (ァリール部分の炭素数 6〜 10で、 アルキル 部分の炭素数 1〜4) 、 又は置換基を有していても良いァリール基若しくは複素 璟基を表し、 Aは酸素原子、 硫黄原子、 又は NR5 (R5は水素原子又は炭素数 1 〜8のアルキル基を表す。 ) を表し、 X1及び X2はそれぞれ独立して結合手、 酸 素原子、 S (0) p (pは 0〜2の整数を表す。 ) 、 C (二 0) 、 C (二 N—〇 6) (R6は水素原子又は炭素数 1〜8のアルキル基を表す。 ) 、 C ( = 0) N H、 NH C ( = 0) 、 S02NH、 NHS〇2, CH (OR7) (R7は水素原子又 は炭素数 1〜8のアルキル基を表す。 ) 、 CH = CH、 又は C≡Cを表し、 Yは 置換基を有していても良い炭素数 1〜 8のアルキレン鎖を表し、 Zは酸素原子又 は硫黄原子を表し、 R3及び R4はそれぞれ独立して置換基を有していても良い炭 素数 1〜 8のアルキル基を表し、 そして は水素原子又は炭素数 1〜 8のアル キル基を表す。
但し、 X1が結合手のとき、 X2は 0、 S (0) pでなく、 また X1が C ( = 0
) NHのとき、 X2は結合手でない。 )
で表される化合物又はその塩に関する。 また本発明は次の一般式 (11) 、
(式中、 R11及び: R12はそれぞれ独立して炭素数 1〜8のアルキル基、 1〜3 個のハロゲン原子で置換された炭素数 1〜 8のアルキル碁、 炭素数 2〜 8のアル ケニル基、 炭素数 2〜 8のアルキニル基、 3〜7員環のシクロアルキル基、 3〜 7員環のシク口アルキル基で置換された炭素数 1〜 8のアルキル基、 又は置換基 としてハロゲン原子、 水酸基、 ニトロ基、 アミノ基、 炭素数 1〜8のアルキル基 、 1〜3個のハロゲン原子で置換された炭素数 1〜8のアルキル基、 炭素数 1〜 8のアルコキシ基、 1〜 3個のハロゲン原子で置換された炭素数 1〜 8のアルコ キシ基、 フエニル基、 ベンジル基、 フエニルォキシ基、 ペンゾィル基若しくはピ リジル基から選ばれる基若しくは原子を有していても良いフヱニルアルキル基 ( アルキル部分の炭素数 1〜4 ) 、 フヱニル基、 ナフチル基、 ピリジル基、 チェ二 ル基、 フリル基、 キノリル基、 ベンゾフラニル基若しくはベンゾチェ二ル基を表 し、 X11及び X12はそれぞれ独立して結合手、 S (0) q (qは 0〜2の整数を 表す。 ;) 、 C (二 0) 、 C (二 N— OR16) (R16は水素原子又は炭素数 1〜8 のアルキル基を表す。 ) 、 C (=〇) NH、 NH C ( = 0) 、 S〇2NH、 NH S〇2, CH (OR17) (R17は水素原子又は炭素数 1〜8のアルキル基を表す 。 ) 、 CH = CH、 又は C三 Cを表し、 Y1は置換基として炭素数 1〜8のアル キル基若しくは炭素数 1〜8のアルコキシ基を有していても良い炭素数 1〜 8の アルキレン鎖を表し、 Z1は酸素原子又は硫黄原子を表し、 そして R13及び R14 はそれぞれ独立して置換基としてハロゲン原子又は炭素数 1〜 8のアルコキシ基 を有していても良い炭素数 1〜 8のアルキル基を表す。
但し、 X11が結合手のとき、 X12は 0、 S (0) pでなく、 ま X11が C (
0) NHのとき、 X 12は結合手でない。 )
で表されるォキサゾ一ル誘導体又はその塩に関する。 また本発明は、 次の一般式 (III) 、
(式中、 : R21及び; R22はそれぞれ独立して炭素数 1〜8のアルキル基、 1〜3個 のハロゲン原子で置換された炭素数 1〜8のアルキル基、 炭素数 2〜 8のァルケ ニル基、 炭素数 2〜8のアルキニル基、 3〜 7員環のシクロアルキル基、 3〜7 員環のシクロアルキル基で置換された炭素数 1〜 8のアルキル基、 又は置換基と してハロゲン原子、 水酸基、 ニトロ基、 アミノ基、 炭素数 1〜8のアルキル基、 1〜 3個のハロゲン原子で置換された炭素数 1〜 8のアルキル基、 炭素数 1〜 8 のアルコキシ基、 1〜 3個のハロゲン原子で置換された炭素数 1〜 8のアルコキ シ基、 フヱニル基、 ベンジル基、 フエニルォキシ基、 ベンゾィル基若しくはピリ ジル基から選ばれる基若しくは原子を有していても良いフエニルアルキル基 (ァ ルキル部分の炭素数 1〜4 ) 、 フヱニル基、 ナフチル基、 ピリジル基、 チェニル 基、 フリル基、 キノリル基、 ベンゾフラニル基若しくはベンゾチェ二ル基を表し 、 X21及び X22はそれぞれ独立して結合手、 S (0) r (rは 0〜2の整数を表す 。 ) 、 C ( = 0) 、 C ( = N-OR26) (R26は水素原子又は炭素数 1〜8のァ ルキル基を表す。 ) 、 C (二 0) NH、 NH C ( = 0) 、 S〇2NH、 NH S 02 , CH (OR27) (R27は水素原子又は炭素数 1〜8のアルキル基を表す。 ) 、 CH = CH、 又は C≡Cを表し、 Y2は置換基として炭素数 1〜8のアルキル基 若しくは炭素数 1〜8のアルコキシ基を有していても良い炭素数 1〜8のアルキ レン鎖を表し、 Z2は酸素原子又は硫黄原子を表し、 そして R23及び; R24はそれ
それ独立して置換基としてハロゲン原子又は炭素数 1〜8のアルコキシ基を有し ていても良い炭素数 1〜 8のアルキル基を表す。
但し、 X21が結合手のとき、 X22は 0、 S (0) rでなく、 また X21が C (= 0) NHのとき、 X22は結合手でない。 )
で表されるチアゾ一ル誘導体又はその塩に関する。 更にまた、 本発明は上記一般式 ( I ) で表される化合物、 上記一般式 ( I I ) で表されるォキサゾ一ル誘導体、 若しくは上記一般式 (III) で表されるチアゾ ール誘導体又はこれらの塩を有効成分として含有するペルォキシゾーム増殖剤応 答性受容体 (5の活性化剤に関する。 発明を実施するための最良の形態
次に本発明を詳細に説明する。
上記一般式 ( I ) における記号の説明をする。
上記一般式 ( I ) .において、 I^、 R R5、 R6、 R7及び R8の炭素数 1〜8 のアルキル基としては、 メチル基、 ェチル基、 プロビル基、 イソプロピル基、 プ チル基、 i—ブチル基、 t—プチル基、 又はペンチル基が挙げられる。
R1及び R 2のハロゲン原子で置換された炭素数 1〜 8のアルキル基としては 1 ~3個のフッ素原子、 塩素原子若しくは臭素原子等のハロゲン原子により置換さ れたメチル,基、 ェチル基、 プロピル基、 イソプロピル基、 プチル基、 または t— ブチル基が挙げられ、 好ましくはトリフルォロメチル基、 クロロメチル基、 2 _ クロ口ェチル基、 2—ブロモェチル基、 2—フルォロェチル基等が挙げられる。
R1及び R2の炭素数 2〜8のアルケニル基としては、 ビニル基、 ァリル基が挙 げられる。
R1及び R2の炭素数 2〜 8のアルキニル基としては、 プロパルギル基が挙げら れる。
R1及び R2の 3〜 7員環のシクロアルキル基としては、 シクロへキシル基、 シ クロペンチル基等が挙げられる。
R1及び R2の 3〜 7員環のシクロアルキル基で置換された炭素数 1〜 8のアル
キル基としては、 シクロへキシルメチル基、 シクロペンチルメチル基等が挙げら れる。
; R 1及び R 2の置換基を有していても良いァリ一ルアルキル基 (ァリ一ル部分の 炭素数 6〜 1 0で、 アルキル部分の炭素数 1〜4 ) としては、 置換基として、 ハロゲン原子 (フッ素原子、 塩素原子、 臭素原子等) 、 水酸基、 ニトロ基、 アミ ノ基、 炭素数 1〜 8のアルキル基 (メチル基、 ェチル基、 プロビル基、 i —プロ ピル基、 ブチル基、 i—ブチル基、 t—プチル基等) 、 1〜3個のハロゲン原子 で置換された炭素数 1〜 8のアルキル基 (トリフルォロメチル基、 ト リフルォロ ェチル基等) 、 炭素数 1〜8のアルコキシ基 (メ トキシ基、 ェトキシ基等) 、 1 〜 3個のハロゲン原子で置換された炭素数 1〜 8のアルコキシ基 ( 2—クロロェ トキシ等) 、 フエニル基、 ベンジル基、 フエニルォキシ基、 ベンゾィル基若しく はピリジル基から選ばれる基又は原子を有していても良いフエニル基又はナフチ ルにより置換された炭素数 1〜 4のアルキル基が挙げられ、 好ましくは炭素数 1 〜 6のアルキル基 (メチル基、 ェチル基、 プロピル基等) 、 炭素数 1〜6のアル コキシ基 (メ トキシ基、 エトキシ基) 又はハロゲン原子 (フッ素原子、 塩素原子 、 臭素原子) 等の置換基を有していても良いフヱニル基で置換されたメチル基、 ェチル基が挙げられ、 さらに好ましくはべンジル基、 ベンズヒ ドリル基、 フエネ チル基等が挙げられる。 及び R 2の置換基を有していても良いァリール基としては、 ハロゲン原子 ( フッ素原子、 塩素原子、 臭素原子) 、 水酸基、 ニトロ基、 アミノ基、 置換アミノ 基 (ジメチルァミノ基) 、 炭素数 1〜 8のアルキル基 (メチル基、 ェチル基、 プ 口ピル基、 i —プロピル基、 プチル基、 i —ブチル基、 t—プチル基) 、 1〜3 個のハロゲン原子で置換された炭素数 1〜 8のアルキル基 (トリフルォロメチル 基、 トリフルォロェチル基) 、 炭素数 1〜 8のアルコキシ基 (メ トキシ基、 ェト キシ基) 、 1〜 3個のハロゲン原子で置換された炭素数 1〜8のアルコキシ基 ( 2—クロ口エトキシ等) 、 ァシル基 (ァセチル基、 ベンゾィル基) 、 カルボキシ ル基、 フエニル基、 ベンジル基、 フヱニルォキシ基若しくはピリジル基等の置換
基を有していても良いフエニル基又はナフチル基が挙げられ、 好ましくはフエ二 ル基、 2—クロ口フエ二ル基、 3 —クロ口フエ二ル基、 4—クロ フエニル基、 2, 3—ジクロロフェニル基、 2 , 4—ジクロロフェニル基、 2, 6—ジクロ口 フエニル基、 2 —フルオロフェニル基、 2 — ト リフルォロメチルフエニル基、 4 —クロ口一 2 —ヒ ドロキシフエニル基、 2 —メチルフエニル基、 4 —プチルフエ ニル基又はナフチル基が挙げられる。
R 1及び R 2の置換基を有していても良い複素環基としては、 環形成原子として 1〜 3個の窒素原子、 酸素原子又は硫黄原子から選ばれるヘテロ原子と残りの炭 素原子からなるピリジル基、 チェニル基、 フ リル基、 チアゾリル基等の 5〜 8員 環の複素環基又はかかる複素環とベンゼン環が縮合したキノ リル基、 ベンゾフラ ニル基又はべンゾチェニル基等が挙げられる。
これらの複素環基は、 上記の R 1及び R 2の置換基を有していても良いァリール 基が有していても良い置換基と同様な置換基で置換されていても良い。
Yは置換基として炭素数 1 ~ 8のアルキル基 (メチル基、 ェチル基、 プロピル 基等) 又は炭素数 1〜 8のアルコキシ基 (メ 卜キシ基、 エトキシ基) を有してい ても良い炭素数 1〜 8のアルキレン鎖であり、 好ましくは炭素数 1〜 6のアルキ レン鎖が挙げられ、 更に好ましくはメチレン、 エチレン、 プロピレンが挙げられ る。
R 3及び R4の置換基を有していても良い炭素数 1〜 8のアルキル基としては、 置換基としてハロゲン原子 (フッ素原子、 塩素原子、 臭素原子等) 又は炭素数 1 〜8のアルコキシ基 (メ トキシ基、 ェトキシ基等) を有していても良い炭素数 1 〜 8のアルキル基が挙げられ、 好ましくはメチル基、 ェチル基、 プロピル基が挙 げられる。 次に上記一般式 ( I I ) における記号の説明をする。
上記一般式 (ェ I ) において、 R "、 R 1 2、 R 1 6及び R 1 7の炭素数 1〜8のァ ルキル基としては、 メチル基、 ェチル基、 プロピル基、 イソプロピル基、 ブチル
基、 i—ブチル基、 t—プチル基、 又はペンチル基が挙げられる
^"及び!^ の 1〜 3のハロゲン原子で置換された炭素数 1へ のアルキル基 としては 1〜 3個のフッ素原子、 塩素原子若しくは臭素原子等のハロゲン原子に より置換されたメチル基、 ェチル基、 プロピル基、 イソプロピル基、 ブチル基、 または t—ブチル基が挙げられ、 好ましくはトリフルォロメチル基、 クロロメチ ル基、 2—クロ口ェチル基、 2—プロモェチル基、 2—フルォロェチル基等が挙 げられる。
R11及び R12の炭素数 2〜8のアルケニル基としては、 ビニル基、 ァリル基が 挙げられる。
R11及び R 12の炭素数 2〜 8のアルキニル基としては、 プロパルギル基が挙げ られる。
R11及び R12の 3〜 7員環のシクロアルキル基としては、 シクロへキシル基、 シクロペンチル基等が挙げられる。
R11及び: R12の 3〜 7員璟のシクロアルキル基で置換された炭素数 1〜 8のァ ルキル基としては、 シクロへキシルメチル基、 シクロペンチルメチル基等が挙げ られる。 また R11及び R12がフエニルアルキル基 (アルキル部分の炭素数 1〜4) 、 フ ェニル基、 ナフチル基、 ピリジル基、 チェニル基、 フリル基、 キノリル基、 ベン ゾフラニル基若しくはベンゾチェニル基の場合、 これらのフヱニル基、 ナフチル 基又は複素環基は置換基としてハロゲン原子 (フッ素原子、 塩素原子、 臭素原子 等) 、 水酸基、 ニトロ基、 アミノ基、 炭素数 1〜 8のアルキル基 (メチル基、 ェ チル基、 プロピル基等) 、 1〜3個のハロゲン原子で置換された炭素数 1〜8の アルキル基 (トリフルォロメチル基、 トリフルォロェチル基等) 、 炭素数 1〜8 のアルコキシ基 (メ トキシ基、 エトキシ基等) 、 1〜 3個のハロゲン原子で置換 された炭素数 1〜 8のアルコキシ基 ( 2—クロ口エトキシ基等) 、 フエニル基、 ベンジル基、 フヱニルォキシ基、 ベンゾィル基若しくはピリジル基から選ばれる 基若しくは原子を有していても良い。 ここでフエニルアルキル基 (アルキル部分
の炭素数 1〜4) としては、 ベンジル基、 ベンズヒ ドリル基又はフエネチル基等 が挙げられる。
Y1は置換基として炭素数 1〜8のアルキル基 (メチル基、 ェチル基、 プロピ ル基等) 若しくは炭素数 1〜8のアルコキシ基 (メ トキシ基、 ェトキシ基) を有 していても良い炭素数 1〜 8のアルキレン鎖であり、 好ましくは炭素数 1〜 6の アルキレン鎖が挙げられ、 更に好ましくはメチレン、 エチレン、 プロピレンが挙 げられる。
R13及び R14は置換基としてハロゲン原子 (フッ素原子、 塩素原子、 臭素原子 等) 又は炭素数 1〜8のアルコキシ基 (メ トキシ基、 エトキシ基) を有していて も良い炭素数 1〜 8のアルキル基が挙げられ、 好ましくはメチル基、 ェチル基、 プロピル基が挙げられる。 次に上記一般式 (III) における記号の説明をする。
—般式 (III) における R21, R22, 23, R 24 5 R26, : R27及び Y2は、 それぞれ上記一般式 ( I I ) の R1 1, R12, R13, R14, R 16, R17及び Y 1と同様なものが挙げられる。
( 1 ) 本発明化合物としては、 上記一般式 ( I I ) で表されるォキサゾール誘 導体で、 X 11が結合手であるォキサゾール誘導体又はその塩が好ましい。
( 2 )また、 本発明化合物としては、 上記一般式 ( I I) で表されるォキサゾ一 ル誘導体又は上記 ( 1 ) で、 が結合手、 c ( = 0) 、 c ( = N-OH) 、 C ( =〇) NH、 NHC (=〇) , CH (OH) 又は C H二 C Hであるォキサゾ一 ル誘導体又はその塩が好ましい。
( 3) また、 本発明化合物としては、 上記一般式 ( I I) で表されるォキサゾ —ル誘導体又は上記 ( 1) 若しくは ( 2) で、 R11が置換基としてハロゲン原子
、 水酸基、 ニトロ基、 アミノ基、 炭素数 1〜 8のアルキル基、 1〜 3個のハロゲ ン原子で置換された炭素数 1〜8のアルキル基、 炭素数 1〜8 ルコキシ基、 1〜 3個のハロゲン原子で置換された炭素数 1〜 8のアルコキシ基、 フエニル基 、 ベンジル基、 フエニルォキシ基、 ベンゾィル基若しくはピリジル基から選ばれ る基若しくは原子を有していても良いフエニル基、 ナフチル基、 ピリジル基、 チ ェニル基、 フリル基、 キノ リル基、 ベンゾフラニル基若しくはベンゾチェ二ル基 であるォキサゾ一ル誘導体又はその塩が好ましい。
(4) また、 本発明化合物としては、 上記一般式 (I I) で表されるォキサゾ —ル誘導体又は上記 (1) 若しくは (2) で、 R11がフエ二ル基、 2—クロロフ ェニル基、 3—クロ口フエ二ル基、 4一クロ口フエ二ル基、 2, 3—ジクロロフ ェニル基、 2, 4ージクロロフェニル基、 2, 6—ジクロロフェニル基、 2—フ ルオロフェニル基、 2 _ トリフルォロメチルフエニル基、 4—クロロー 2—ヒ ド ロキシフエニル基、 2—メチルフエニル基、 4一プチルフエニル基又はナフチル 基であるォキサゾ一ル誘導体又はその塩が好ましい。
(5) また、 本発明化合物としては、 上記一般式 (I I) で表されるォキサゾ —ル誘導体又は上記 (1) 〜 (4) で、 R12が炭素数 1〜8のアルキル基又は 1 〜 3個のハロゲン原子で置換された炭素数 1〜 8のアルキル基であるォキサゾ一 ル誘導体又はその塩が好ましい。
(6) また、 本発明化合物としては、 上記一般式 (I I) で表されるォキサゾ —ル誘導体又は上記 ( 1) 〜 (5) で、 R11の置換位置がォキサゾ一ル環の 2位 であるォキサゾール誘導体又はその塩が好ましい。
(7) また、 本発明化合物としては、 上記一般式 (III) で表されるチアゾ一 ル誘導体で、 X21が結合手であるチアゾ一ル誘導体又はその塩が好ましい。
(8)また、 本発明化合物としては、 上記一般式 (III) で表されるチアゾ一ル
誘導体又は上記 (7) で、 X22が結合手、 C ( = 0) 、 C ( = N— OH) 、 C ( =0) NH、 NH C ( = 0) , CH (OH) 又は CH = CHで チアゾ一ル誘 導体又はその塩が好ましい。
(9) また、 本発明化合物としては、 上記一般式 (III) で表されるチアゾ一 ル誘導体又は上記 (7) 若しくは (8) で、 R21が置換基としてハロゲン原子、 水酸基、 ニトロ基、 アミノ基、 炭素数 1〜 8のアルキル基、 1〜 3個のハロゲン 原子で置換された炭素数 1〜8のアルキル基、 炭素数 1〜8のアルコキシ基、 1 〜 3個のハロゲン原子で置換された炭素数 1〜 8のアルコキシ基、 フェニル基、 ベンジル基、 フエニルォキシ基、 ベンゾィル基若しくはピリジル基から選ばれる 基若しくは原子を有していても良いフエニル基、 ナフチル基、 ピリジル基、 チェ ニル基、 フリル基、 キノ リル基、 ベンゾフラニル基若しくはベンゾチェ二ル基で あるチアゾ一ル誘導体又はその塩が好ましい。
(10) また、 本発明化合物としては、 上記一般式 (III) で表されるチアゾ —ル誘導体又ば上記 (7) 若しくは (8) で、 R21がフヱニル基、 2—クロロフ ェニル基、 3—クロ口フエ二ル基、 4—クロ口フエ二ル基、 2, 3—ジクロロフ ェニル基、 2 , 4—ジクロロフェニル基、 2, 6—ジクロロフェニル基、 2—フ ルオロフェニル基、 2—トリフルォロメチルフエニル基、 4—クロ口一 2—ヒド ロキシフエニル基、 2—メチルフエニル基、 4一ブチルフエニル基又はナフチル 基であるチアゾ一ル誘導体又はその塩が好ましい。
(1 1) また、 本発明化合物としては、 上記一般式 (III) で表されるチアゾ —ル誘導体又は上記 (7) 〜 ( 10) で、 R22が炭素数 1〜8のアルキル基又は 1〜 3個のハロゲン原子で置換された炭素数 1〜 8のアルキル基であるチアゾ一 ル誘導体又はその塩が好ましい。 .
( 12) 更にまた、 本発明化合物としては、 上記一般式 (III) で表されるチ ァゾール誘導体又は上記 (7) 〜 (1 1) で、 R21の置換位置がチアゾール環の
2位であるチアゾール誘導体又はその塩が好ましい。 上記一般式 (I) で表される本発明化合物にはシス、 トランスの幾何異性体や 光学異性体等も存在する場合もあるが、 これらの異性体も本発明に含まれる。 更にまた、 本発明化合物としては、 ナトリウム塩、 カリウム塩等のアルカリ金 属塩等の製薬学的に許容される塩も含まれる。 次に本発明化合物である一般式 (I) の製造方法を記載する。
合成方法 1
(a) (b)
(式中、 Qはトシルォキシ基、 ハロゲン原子 (臭素原子等) 等の脱離基を表し そして R R2, R3, R4、 R8, A、 X1, X2, Y及び Zは前記と同じ。 ) 上記製法において、 本発明化合物である一般式 (I) で表される化合物は、 一 般式 (a) で表されるフエノール又はチオフヱノール化合物と一般式 (b) で表 される酢酸誘導体を反応させることにより得ることができる。
反応は、 ェチルメチルケトン等の溶媒中、 炭酸カリウム等の塩基の存在下行う ことができる。 尚、 原料である一般式 (a) で表されるフヱノール又はチオフヱノール化合物 は、 例えば以下で示す合成スキームと同様な方法を用いて製造できる。
<原料合成例 1 >
(1)
F EttnO 1入^ IOEt ,NaH I EtjO- THF
2
1)
,ΟΒπ R CH2' ,画/ THF R2
Et02C R 、
2) c HC1 / AcOH
(2)
(式中、 Bnはベンジル基を表し、 そして R1, R2及び Aは前記と同じ。 ) また、 下記の原料合成例 2, 3記載のスキーム等を用いて製造できる。
<原料合成例 2 >
2)脱炭酸、
基の脱保護
R2 脱水 垤兀
R1 (CHa) π+1
OHG- ow
A=0,S,NH
<原料合成例 3 >
R=H,Me etc
W=保護基
A=0,S,NH
(式中、 B nはベンジル基を表し、 h a 1 oはハロゲン原子を表し、 nは整数 を表し、 Wはベンジル基等の保護基を表し、 そして R 1 , R 2は前記と同じ。 ) 上記原料合成例 2において、 2-ベンゾィル酢酸ェチル誘導体とハロゲン誘導体 とを縮合した後、 得られたケトン体を脱炭酸、 ベンジル基の脱保護を行いァシル 置換されたフヱノ一ル体を得る。 このァシル置換されたフヱノ一ル体を N a B H 4 , L i A 1 H4等の還元剤を用い ( 1—ヒドロキシアルキル) フエノール体に変 換出来る。 この ( 1—ヒドロキシアルキル) フヱノール体は硫酸等の酸性条件下
やハロゲン化試剤、 スルホン化試剤、 脱水剤を用いることでォレフィン置換フエ ノール体に変換出来る。 尚、 ォレフィン体はべンズアルデヒ ド类 Wi t t i g 試薬と作用させることにより得ることもできる。 これらのォレフィ ン置換フエノ —ル体にエタノール等の溶媒中、 P d— C等の触媒存在下にて接触還元反応を行 うことでアルキル置換フエノール体を得ることができる。
また、 原料合成例 3に記載している方法により、 ァシル置換されたフヱノール 体からォキシム体を得ることができ、 さらにアミン類と安息香酸クロリ ド誘導体 を反応させることで (保護基を有する場合はさらに脱保護することで) カルバモ ィル置換フエノール体を、 またァシルクロリ ドとァ二リン類を反応させることで (保護基を有する場合はさらに脱保護することで) ァシルァミノ置換フヱノール 体を得ることができる。 合成方法 2
(c)
(式中、 I まメチル基、 ェチル基等の炭素数 1〜 6のアルキル基を表し、 そし て R1, 2, R3, R4、 A、 X1, 2, Y及び Zは前記と同じ。 ) 上記製法において、 本発明化合物である一般式 (I) で表される化合物 (R8
=水素原子) は、 一般式 ( C ) で表されるエステル体をエタノール—水等の溶媒 中、 水酸化ナトリウム、 水酸化カリウム、 水酸化リチウム等の の存在下、 加 水分解反応に付すことで得ることができる。 合成方法 3
(e)
(式中、 Υ
ϋは炭素数 1〜6のアルキレン鎖又は結合手を表し、 そして R
1 , R
3, R
4、 R
8, A、 X
1及び Zは前記と同じ。 ) 上記製法において、 本発明化合物である一般式 (I) で表される化合物 (X
2 =結合手) は、 一般式 (e) で表されるォレフィ ン体をエタノール中、 Pt—C 等の触媒の存在下、 還元反応に付すことで得ることができる。 合成方法 4
(g)
(式中、 R1, R2, R3, R4、 R8, A、 X1、 Y及び Zは前記と同じ。 ) 上記製法において、 本発明化合物である一般式 (I) で表される化合物 (X2 が C ( = N_OH) ) は、 一般式 (g) で表されるケトン体にヒ ドロキシルアミ ンを作用させることで得ることができる。 かく して得られた本発明化合物の代表化合物例を次に示す。
( 1 - 1) 次の一般式 (I— a) で表される化合物 :
即ち、 上記一般式 (I) で表される化合物で、 A=0、 Z=0、 R8 = Hで、 X1がォキサゾ一ル環の 4位に結合している本発明化合物
(1-2) 上記一般式 (I— a) で表される化合物
【表 2】
(2) 次の一般式 (I一 b) で表される化合物:
即ち、 上記一般式 (I) で表される化合物で、 A=0、 Z = S R8 = Hで、 X 1がォキサゾ一ル環の 4位に結合している本発明化合物
(3- 1) 次の一般式 (I— c) で表される化合物:
即ち、 上記一般式 (I) で表される化合物で、 A≠0、 R3=R4 =メチル、 Z =0、 R8 = Hで、 X1が例えばチアゾ一ル環の場合 4位に結合している本発明化 合物
(3-2) 上記一般式 (I— c) で表される化合物
【表 5】
(4- 1) 次の一般式 (I一 d) で表される化合物:
即ち、 上記一般式 (I) で表される化合物で、 R
3 = R
4 =メチル、 Z二 0、
8 = Hで、 X
1が例えばォキゾ一ル環の場合 4位に結合している本発明化合物
(i一
(4-2) 上記一般式 (I一 d) で表される化合物:
【表 7】
(5- 1) 次の一般式 (I一 e) で表される化合物:
即ち、 上記一般式 (I) で表される化合物で、 R
3 = R
4 =メチル、 Z = 0、 R
8 = Hで、 X
1が例えばチアゾール環の場合 5位に結合している本発明化合物
【表 8】
R1 R2 A X1 Y X2、その 置換位置
(2-CI)フ;!:ニル イソプロピル s 結合手 CH2CH2 C=0(4)
(4 - CI)フ Iニル プロピル S 結合手 CH2CH2 C=0(4)
(2-CI)フ Iニル イソプロピル NH 結合手 CH2CH2 C=N-OH(4)
(2-CI)フヱニル イソプロピル N - Me 結合手 CH2CH2 CH(OH)(4)
(2 - CI)フエニル イソプロピル N-Et 結合手 CH rl2CH2 結合手 (4)
(2 - CI)フヱニル イソプロピル N-Bn 結合手 CH2CH9 CH=CH(4) フエニル メチル S 結合手 CH2CH2 C=0(4)
2-ピリジル ェチル S 結合手 CH2CH2 C=0(4)
2-ナフチル プロピル NH 結合手 結合手 (4) シクロへキシル ブチル N-Me 結合手 CH2CH2 NHC=0(4)
(2-CI)フ; cニル プロピル N-Et 結合手 CH2CH2 CH=CH(4)
(2 - CI)フエニル プロピル N-Bn 結合手 CH2CH2 CH(OH)(4)
(2,4- CI)フヱニル プロピル S 結合手 CH2CH2 C=N-OH(4)
(4- Bu)フエニル へキシル NH 結合手 v_»H2CH2 H2 C=0(4)
(4-tBu)フエニル へキシル N-Me 0 CH2CH2 0(3)
(2 - CI)フエニル t一プチル N-Et NHCO CH2CH2 C=0(3)
(5— 2) 上記一般式 (I一 e) で表される化合物 【表 9】
(6) 次の一般式で表されるォキサゾール誘導体:
(式中、 R1, R2、 R4, R5、 X1, X2及び Y1は表 10〜15記載のもの (
【表 1 0】
R1 R2 R4 R5 X1 Υ1 置換 位置)
(2-Cl)フ エニル イ ソブロビル メ チル メ チル ^合 CH2NH C=0(4)
(2 - CI)フ ユニル イ ソ ブ σビル メ ル メ チル ^合 CH2C=0 NH(4)
(2— CI)フ ニル イ ソブロビル メ チル メ ル ίま会寻 *-»Hy,CH2Ch2 C=0(4)
(2, 4-C1)フ ユニル イ ンブロビル メ チル メ チル ^合寻 ^H->CH2 C=0(4)
(2, 4-C1)フ ユニル ィ ン ブロビル メ チル ェチル ί 合寻 H2し H2 C=0(4)
(3,4— C1)フ ユニル イ ソブロピル メ チル メ チル ίま合 CH2CH2 C=0(4) -ベ ンゾフ ラニル イ ソ ブロビル メ チル メ チル ま合ネ CH2CH2 C=0(4)
4 ビフ ユ二 リ ル イ ソブロビル メ チル メ チル 会寻 CH2CH2 C=0(4)
1— 0H-2—ナフチル イ ソブロビル メ チル メ チル ^合寻 CH2CH C=0(4)
(2, 4-C1)フ -ル ィ ン ブロビル メ チル メ チル ^合寻 CH2 CH=CH(4) - 0H - 2 -ナフチル ィ ソ ブロ ピル メ チル メ チル ί 合 CH2CH2 C=0(4)
(2 - 0H,4- C1)フ -ル イ ソブロピル メ チル メ チル 合寻 CH2CH2 C=0(4)
(2— C1, 4— Br)フ ユニル ィ ソ ブロピル メ チル メ チル i合 CH2CH C=0(4) — C1— 4 ビフ ェニリ ル イ ソブロピル メ チル メ チル 合ネ CH2CH2 C=0(4)
(2 - 0H,4— Me)フ ユニル イ ソブロビル メ 手ル メ チル ^合 CH2CH2 C=0 (4)
(2, 4- Me)フ エニル イ ソブロピル メ チル メ チル ί 合 CH2CH2 C=0(4)
(2 - 0H)フ ヱニル イ ンブロビル メ チル メ チル ^合 CH2CH2 C=0(4)
(2-0H,3,4- e)フ エニル イ ソブロビル メ チル メ チル ^合ネ CH2CH2 C=0(4)
(2-OH, 4-CF3)フ ユニル ィ ソ ブ σビル メ チル メ チル ^合ネ CH2CH2 C=0(4)
(2 - Cl,4 - OMe)フ エニル ィ ソ ブロ ピル メ チル メ チル ネ CH2CH2 C=0(4)
【表
R1 R2 R4 R5 X1 Υ1 x2(置換位置)
1 -ナフチル ブ σビル メ チル メ チル .iま合ネ CH2CH2 C=0(4)
2-ナフチル ブ口ビル メ チル メ チル ^合ヰ CH2CH2 C=0(4)
2—キ ノ ') ル ブ口ピル メ チル メ チル i 合寻 CH2CH2 C=0(4)
8—キ ノ リ ル ブ口ピル メ チル メ チル 合斗 CH2CH2 C=0 (4)
3—キ ノ リ ル ブ口 ビル メ チル メ チル ^合 CH2CH2 C=0(4)
2 -ビ ') ミ シ'ル ブ σ ピル メ チル メ チル iま合寻 CH2CH2 C=0(4)
2-チェニル ブ口ビル メ チル メ チル . 合寻 CH2CH2 C=0(4)
2—フラ ュル ブ口ビル メ チル メ チル ίま合寻 CH2CH2 C=0(4)
2 -ィ ミ ダゾリ ル ブ口ビル メ チル メ チル . 合寻 CH2CH2 C=0(4) —ィ ンド リ ル プ口ビル メ チル メ チル i 合 4 CH2CH2 C=0 (4)
-";ンソ'チェニル ブ口ピル メ チル メ チル .ま合寻 CH2CH2 C=0 (4)
-べンズイ ミ ダゾリ ル ブ口ビル メ チル メ チル 、 合ネ CH2CH2 C=0(4)
(2-C1)フ ユュル イ ソ ブロ ピル メ チル メ チル iま合寻 CH2NH C=0(3)
(4-C1)フ ニル ィ ソブロビル メ チル メ チル 合寻 CH2C=0 NH(3)
(2-C1)フ ユニル ィ ソブロ ピル メ チル メ チル ^合 CH2CH2CH2 C=0(3)
(2,4-Cl)フ ユニル ィ ソブロビル メ チル メ チル ^合 4 CH2CH2 C=0(3)
(2,4-Cl)フ -ル イ ソブロピル メ チル ェチル . 合寻 CH2CH2 C=0 (3)
(3,4-Cl)フ ユ -ル ィ ソブロピル メ チル メ チル . 合寻 CH2CH2 C=0 (3)
-べンゾフ ラニル イ ソブロ ピル メ ル メ チル iま合寻 CH2CH2 C=0 (3)
4一ビフ エ - リ ル イ ソブロビル メ チル メ チル 含 4 CH2CH2 C=0(3)
【表 13】
R1 R2 R4 R5 X1 Υ1 (置換 位置)
1- OH-2-ナフ チル ィ ソブ口ビル メ チル メ チル . 合寻 し h CH2 C=0(3)
(2, 4- CI)フ エ ニル ィ ソブロビル メ チル メ チル ίま合寻 CH2 CH=CH (3)
3— OH— 2—ナフ チ ィ ソブロビル メ チル メ チル ^佥ネ CH2CH2 C=0 (3)
(2-0H.4-C1)フ エ ニル ィ ソブロビル メ チル メ チル ^合寻 CHn H2 C=0 (3)
(2 - C1, 4 - Br)フ エニル ィ ソブロビル メ チル メ チル .i 合寻 CH2CH2 C=0{3) -C1- 4 -ビフ エユリ ル イ ソブロビル メ チル メ チル ^合 4 CH2CH2 C=0 (3)
(2-OH, 4-Me)フ エニル ィ ソブロピル メ チル メ チル . 合ネ CH2CH2 C=0 (3)
(2, 4- e)フ エ ニル イ ソプロビル メ チル メ チル ^合寻 CH2CH2 C=0 (3)
(2-0H) フ エ ニル ィ ソブロピル メ チル メ チル . 合 CH2CH2 C=0 (3)
(2-0H, 3, 4- e)フ エニル ィ ソブロピル メ チル メ チル 合寻 CH2CH2 C=0 (3)
(2-0H.4-CF3)フ エニル ィ ソブロビル メ チル メ チル ^佥 CH2CH2 C=0 (3)
(2-Cl, 4-OMe)フ エニ レ ィ ソブロビル メ チル メ チル iま合ヰ CH2CH2 C=0(3)
(2 - C1, 4- OPh)フ エニル イ ソプロピル メ チル メ チル 合 4 CH2CH2 C=0 (3)
1-ナフチル イ ソブロピル メ チル メ チル ^会寻 CH2CH2 C=0 (3) -ナフチル ィ ソブロピル メ チル メ チル iま合寻 CH2CH2 C=0 (3) —キ ノ リ ル イ ソ プロビル メ チル メ チ ·>レ . 合寻 CH2CH2 C=0 (3) キ ノ 1 J ル イ ソブロビル メ チル メ チル ^会斗 CH CH2 0 (3)
3 キ ノ リ ル ィ ソブロピル メ チル メ チル .ま合寻 CH2CH2 C=0 (3) -ビ ') ミ シ"ル イ ソプロピル メ チル メ チル 合 CH2CH2 C=0(3)
【表 1 4 】
(7) 次の一般式で表されるォキサゾール誘導体 (
(式中、 R1, R2、 R4, R5、 X1, X2及び Y1は表 16〜21記載のもの,
【表 1 6】
R1 2 4 5 X1 Υ1 X2( 〖位置)
(2-Cl)フ エニル ィ ソ ブロビル メ チル メ チル i 合寻 CH2NH C=0(4)
(2 - CI)フ ユニル ィ ソプロビル メ チル メ チル iま合ヰ CH2C=0 NH(4)
(2-C1)フ ユ -ル ィ ソ ブロビル メ チル メ チル 合寻 CH2CH2CH2 C=0(4)
(2, 4-C1)フ ユ -ル ィ ソ ブロビル メ チル メ チル . 含寻 CH2CH2 C=0(4)
(2, 4-C1)フ エ エル ィ ソブロピル メ チル 工 ル ^合ネ CH2CH2 C=0(4)
(3, 4-C1)フ ユニル ィ ソ ブロビル メ チル メ チル CH2CH2 C=0(4) 一":ンゾ 'フ ラ二ル イ ソ ブロ ピル メ チル メ チル iま合寻 CH2CH2 C=0(4)
4ービフ エ二リ ル イ ソブ σピル メ チル メ チル iま合寻 CH2CH2 C=0(4)
1- 0H-2-ナフチル ィ ソブロビル メ チル メ チル ^合寻 CH2CH2 C=0(4)
(2, 4-C1)フ ユ -ル ィ ソ ブロビル メ チル メ チル ^合 4 CHZ CH=CH{4) - 0H - 2 -ナフチル ィ ソ ブロビル メ チル メ チル ま合 4 CH2CH2 C=0(4)
(2 - 0H,4 - C1)フ ユニル ィ ソブロピル メ チル メ チル ίま合ネ CH2CH2 C=0(4)
(2-Cl,4-Br)フ ユニル ィ ソ ブロビル メ チル メ チル iま合ネ CH2CH2 C=0(4) -C1-4 -ビフ ェニ リ ル イ ソブ σビル メ チル メ チル ま合寻 CH2CH2 C=0(4)
(2-0H,4-Me)フ ユニル ィ ソ ブロピル メ チル メ チル ^合ネ CH2CH2 C=0 (4)
(2,4- Me)フ ユ -ル ィ ソ ブロピル メ チル メ チル .iま合寻 CH2CH2 C=0(4)
(2-0H)フ ユニル ィ ソブロピル メ チル メ チル ί 合 4 CH2CH2 C=0(4)
(2 - 0H, 3, 4 - Me)フ ユニル ィ ソブロビル メ チル メ チル 含寻 CH2CH2 C-0(4)
(2-0H,4-CF3)フ エ -ル ィ ソ ブロピル メ チル メ チル 、 合寻 CH2CH2 C=0 (4)
(2-Cl,4-0 e)フ エニル イ ソブロビル メ チル メ チル . 合寻 CH2CH C=0(4)
【表 1 7】
R1 R2 . R4 R5 X1 Y1 !置換 位置)
(2-Cl,4-0Ph)フ エニル イ ソブ σ ピル メ チル メ チル 合ヰ C=0(4)
1 -ナフチル ィ ソブ口ビル メ チル メ チル ヰ CH2CH2 C=0(4)
2-ナフチル ィ ソブロ ビル メ チル メ チル i 合寻 CH2CH2 C=0(4)
2—キ ノ 1) ル ィ ソブロ ビル メ チル メ チル . 合ネ CH2CH2 C=0(4)
8—キ ノ リ ル ィ ソブロ ビル メ チル メ チル . 合斗 CH2CH2 C=0(4)
3—キ ノ リ ル ィ ソブロ ビル メ チル メ チル 合 4 CH2CH2 C=0(4)
2 -ビリ ミ シ'ル ィ ソブロ ピル メ チル メ チル ま合寻 CH2CH2 C=0(4)
2-チェニル ィ ソブロ ピル メ チル メ チル ま含ヰ CH2CH2 C=0(4)
2-フラニル イ ソブロピル メ チル メ チル 結合 4 CH2CH2 C=0(4)
2-ィ ミ ダゾリ ル ィ ソブロ ピル メ チル メ チル i 合ネ CH2CH2 C=0(4)
2 イ ン ド' 'J ル イ ソブロビル メ チル メ チル 合寻 CH2CH2 C=0(4)
2 -"" ΐンゾチェニル イ ソブ口ビル メ チル メ チル 合斗 CH2CH2 00(4) -ベンズィ ミ ダゾリ ル イ ソブ口ビル メ チル メ チル 合ヰ CH2CH2 C=0(4)
(2-0H,4-Me)フ エニル ブ口ビル メ チル メ チル . 合ネ CH2CH2 O0(4)
(2,4-Me)フ エニル ブ口ビル メ チル メ チル ί 合ネ CH2CH2 00(4)
(2-0H)フ ユニル ブ口ビル メ チル メ チル 合寻 CH2CH2 C=0(4)
(2-0H,3,4-Me)フ ニル ブ口ピル メ チル メ チル 合ネ CH2CH2 C=0(4)
(2-0H.4-CF3)フ ユニル ブ口ピル メ チル メ チル 合ネ CH2CH2 C=0(4)
(2-Cl,4-0 e)フ ユニル ブ口ピル メ チル メ チル ίま合斗 CH2CH2 C=0(4)
(2-Cl,4-0Ph)フ エニル ブ口ビル メ チル メ チル iま合斗 CH2CH2 C=0(4)
【表 1 8 】
(8) 次の一般式で表されるチアゾ一ル誘導体:
即ち、 上記一般式 (I) で表される化合物で、 A=S、 Z = Sである本発明化 合物
【表 22】
(9) 次の一般式で表されるチアゾール誘導体:
即ち、 上記一般式 (I) で表される化合物で、 A=S Z=0である本発明化 合物
(式中、 R1, R R4, R5、 X1, X2及び Y1は表 2 3及び 24記載のもの
)
【表 23】
R1 R2 R4 R5 X1 Υ1 : 置換
H
位置)
( -CF3 フ ユ -ル ブ σピル メ チル メ チル 寻 CH2CH2 C=0(4)
( 4 -Me)フユニル ブ口ピル メ チル メ チル 斗 CH2CH2 C=0(4)
( 4 -OMe)フ ユニル ブチル メ チル メ チル 合斗 CH2CH2 C=0(4)
(4-OPh)フ ユニル キ シル メ チル メ チル 合斗 C=0(4)
(4-0CF3)フ ユニル ィ ソブロビル メ チル ェチル ヰ CH2CH2 C=0(4)
ブ口ビル メ チル メ チル 斗 CH2CH2 C=0(4)
(3- Me)フ ユニル ブチル メ チル メ チル ίま合ネ CH2 C=0(4)
(3 - C1)フ ユ -ル キ シル メ チル メ チル . 合寻 CH2CH2 C=0(3)
(3, 4-OMe)フ ヱニル ィ ソブロビル メ' ル メ チル iま合寻 CH2CH2 C=0(4)
(3,4-Me)フ ユニル ブ口 ピル メ チル メ チル . 合寻 CH2 CH=CH (4)
(3,4-Cl)フ ユ -ル ブチル メ チル ェチル 合寻 GH2CH2 C=0(4)
(2 4 - Me)フ エニル キ シル メ チル メ チル 合斗 CH2CH2 C-0(4) ,4-Cl)フ ユニル ィ ソブロピル メ チル メ チル iま合寻 CH2CH2 C=0(4)
(2-OH, 3,4-Me)フ ユ -ル ィ ノブ口 ビル メ チル メ チル 合ヰ CH2CH2 C=0(4)
(2,4-F)フ ニル イ ソ ブロ ピル メ チル メ チル . 合斗 CH2CH2 C=0(4)
(3, 4, 5 - Me)フ エニル イ ソブロビル メ チル メ チル 合斗 CH2CH2 C=0(4)
(2-OH, 3,4-Me)フ エニル イ ソブロピル メ チル メ チル 合寻 CH2CH2 C=0(4)
次に本発明の薬理効果について述べる。
本発明化合物の P PAR (5活性化作用は、 C V— 1細胞にキメラ受容体発現プ ラスミ ド (GAL 4— hPPAR5 LBD) 、 レポ一夕一プラスミ ド (UAS X4-TK-LUC) 及び 3—ガラク トシダ一ゼ ( ? GAL) 発現プラスミ ド をリポフエクション試薬 D MR I E - C (L i f e Te chno l o gi e s ) により トランスフエク ト後、 本発明化合物又は比較化合物である L— 1650 41の存在下、 40時閭培養後、 可溶化細胞をルシフェラーゼ活性及び/? GA
L活性を測定することにより求めた。
尚、 ルシフェラ一ゼ活性は 5— GAL活性で補正し、 L— 1 6 04 1で処理 した細胞のルシフェラ一ゼ活性を 1 0 0 %として、 相対的なリガンド活性を算出 した。 同様な方法により P P ARひ及びァ活性化作用に関する相対的なリガンド 活性を算出した。 (後記実施例 9 ) 表 2 5記載の様に、 本発明化合物 (実施例 1〜 6) は L— 1 6 5 04 1に比べ 、 これと同等又はそれ以上の PPAR5活性化作用を示し、 また実施例 1及び 5 記載の本発明化合物等については、 P PAR α及びァ活性化作用に比べ、 Ρ ΡΑ R 5に対し選択性の高い活性化作用を示した。
同じく表 2 6記載の様に本発明化合物 (実施例 7— 6等) は L— 1 6 5 0 4 1 に比べ、 これと同等又はそれ以上の P PAR d活性化作用を示し、 また本発明化 合物 (実施例 7— 1 2等) については、 P PARひ及びァ活性化作用に比べ、 P PAR6に対し選択性の高い活性化作用を示した。
また表 2 7記載の様に本発明化合物 (実施例 8—:! 〜 8— 4) は L— 1 6 5 0 1に比べ、 これと同等又はそれ以上の P PARd活性化作用を示した。
従って、 本発明の一般式 ( I ) で表される化合物は、 優れた P PARd活性化 作用を有することから、 血糖降下剤、 脂質低下剤、 肥満、 シンドローム X, 高コ レステロール血症、 高リポ蛋白血症等の代謝異常疾患、 高脂血症、 動脈硬化症、 循環器系疾患、 過食症、 虚血性疾患、 肺ガン、 乳がん、 結腸ガン、 大腸ガン、 卵 巣ガン等の悪性腫瘍、 アルツハイマー病、 炎症性疾患、 骨粗鬆症 (Man o H . e t . A 1. , ( 2 0 0 0 ) J . B i o l . C h e m. , 2 7 5 : 8 1 2 6 - 8 1 3 2) 、 バセドウ病眼症, 副腎白質ジストロフィー等の予防、 あるいは治療 剤として期待される。 本発明化合物は、 ヒトに対して一般的な経口投与又は非経口投与のような適当 な投与方法によつて投与することができる。
製剤化するためには、 製剤の技術分野における通常の方法で錠剤、 顆粒剤、 散 剤、 カプセル剤、 懸濁剤、 注射剤、 坐薬等の剤型に製造することができる。
これらの調製には、 通常の賦形剤、 崩壊剤、 結合剤、 滑沢剤、 素、 希釈剤な どが用いられる。 ここで、 賦形剤としては、 乳糖、 D—マンニト ル、 結晶セル ロース、 プドウ糖などが、 崩壊剤としては、 デンプン、 カルボキシメチルセル口 ースカルシウム (CMC— C a) などが、 滑沢剤としては、 ステアリン酸マグネ シゥム、 タルクなどが、 結合剤としては、 ヒドロキシプロピルセルロース (HP C) 、 ゼラチン、 ポリビニルピロリ ドン (P VP) などが挙げられる。 投与量は通常成人においては、 注射剤で有効成分である本発明化合物を 1日約 0. l mg〜: L O Omg, 経口投与で 1日 1 mg〜 2 0 0 0 mgであるが、 年齢 、 症状等により増減することができる。 次に、 実施例を挙げ本発明を更に詳細に説明するが本発明はこれらに限定され るものではない。 実施例
実施例 1
2 - [4一 [3— [2— ( 2—クロ口フエニル) _ 5—イソプロピル一 4ーォキ サゾリル] プロピオニル] フエニルォキシ] 一 2—メチルプロピオン酸
( 1 ) 4― [ 3一 [ 2 - ( 2—クロ口フエニル) — 5—イソプロピル— 4ーォキ サゾリル] プロピオニル] フエノール 氷冷した T HF ( 5 mL) に 6 0 %水素化ナトリウム (40 mg, 1. 0 0 mm o 1) を加えた。 続いて 2— [ ( 4—ベンジルォキシ) ベンゾィル] 酢酸ェチル
( 3 0 O m g, 1. O Ommo l ) の THF ( 5 mL) 溶液を 3 0分間で滴下し た。 室温に戻し 3 0分攪拌した後、 4—ョードメチルー 5—イソプロピル一 2 ―
( 2—クロ口フエニル) ォキサゾ一ル (3 6 2mg, 1. 0 0 mm o 1 ) を加え た。 窒素雰囲気下にて 20時間加熱還流した後、 室温に戻した。 TH Fを減圧留 去し、 残渣に酢酸 ( 3. OmL) —濃塩酸 ( 0. 8mL) を加え 5時間加熱還流
した。 室温に戻した後、 反応溶液を氷冷水に注ぎ、 酢酸ェチルを加え抽出した。 有機層を分取後、 飽和の炭酸水素ナトリウム水溶液、 水、 食塩水 洗浄し無水硫 酸ナトリウムで乾燥、 濾取した。 酢酸ェチルを減圧留去後、 残渣をシリカゲル力 ラムクロマトグラフィー (へキサン/酢酸ェチル = 3/1 ) にて精製し上記の標 題化合物を微黄白色結晶 (23 Omg) として得た。 (収率 65%)
1 H-NMR (CDC 1 40 OMH z)
δ :
1. 32 (d, 6 H, J = 7H z) ,
2. 96 ( t, 2 H, J = 7 H z ) ,
3. 15-3. 30 (m, 1 H) ,
3. 27 (t, 2H, J = 7 H z) ,
6. 78 (d, 2H, J-8Hz) ,
7. 1 - 7. 2 (b r , 1 H) ,
7. 3 - 7. 4 (m, 2 H) ,
7. 45 - 7. 50 (m, 1 H) ,
7. 79 (d, 2 H3 J二 8Hz)
7. 90 - 7. 95 (m, 1 H)
(2) 2— [4— [3— [2— (2—クロロフヱニル) 一 5—イソプロピル一 4 —ォキサゾリル] プロピオニル] フエニルォキシ] — 2—メチルプロピオン酸ェ チル 上記 ( 1 ) で得られたフエノール化合物 (220mg, 0. 59mmo l) 、 2—プロモー 2—メチルプロビオン酸ェチル ( 348 mg, 1. 78 mm o 1 ) 、 炭酸カリウム ( 246 mg, 1. 78 mmo 1 ) をメチルェチルケトン (5m L) に懸濁させた後、 20時間加熱還流した。 室温に戻した後、 不溶物を濾過、 更にメチルェチルケトンにて洗浄し溶媒を留去した。 この残渣をシリカゲルカラ ムクロマトグラフィー (へキサン/酢酸ェチル =4/1) で精製し上記の標題化 合物を無色油状物 (23 Omg) として得た。 (収率 81 %)
1 H-NMR ( CD C 1 3 , 40 OMH z)
ό1 :
1. 20 (t , 3 H, J = 7 H z ) ,
1. 30 (d, 6 H, J = 7 H z) ,
1. 64 ( s , 6 H) ,
2. 9 6 (t , 2 H5 J = 7 H z) 3
3. 1 0 - 3. 2 5 (m, 1 H) ,
3. 34 (t , 2 H, J = 7 H z ) ,
4. 2 1 (q, 2 H, J = 7 H z ) ,
6. 8 1 (d5 2 H, J = 8 H z ) ,
7. 3 - 7. 4 (m, 2 H) ,
7. 45 - 7. 5 0 (m, 1 H) ,
7. 9 1 (d, 2 H , J二 8 H z) ,
7. 9 0 - 7. 9 5 (m, 1 H)
3) 2 - [4 - [3 - [2 2—クロ口フエニル) 一 5—イソプロピル一 4 一才キサゾリル] プロピオニル] フエニルォキシ] - 2—メチルプロピオン酸 上記 ( 2 ) で得られたエステル体 ( 2 2 Omg, 0. 4 5 mmo 1 ) をェタノ一 ル—水 ( 6mL— 3mL) の混合溶媒に溶解させた後、 水酸化リチウム一水和物 (4 O mg) を加えた。 室温にて 20時間攪拌後、 反応溶液に氷、 希塩酸を加え .中和し酢酸ェチルにて抽出した。 有機層を分取後、 水、 食塩水で洗浄し無水硫 酸ナト リウムで乾燥、 濾取した。 酢酸ェチルを減圧留去し残渣に無色ァモルファ スである標題化合物を 150mg得た。 (収率 7 2 %)
^- MR ( CD C 13 , 40 0 MH z)
(J :
1. 3 1 (d, 6 H, J = 7 H z) ,
1. 65 ( s , 6 H) ,
2. 9 6 (t , 2 H, J = 7 H z) ,
3. 1 5 - 3. 3 0 (m, 1 H) ,
3. 2 8 ( , 2 H, J = 7 H z) ,
6. 8 8 (d, 2 H, J = 8 H z ) ,
7. 3 - 7. 4 (m, 2 H) ,
7. 4 5— 7. 5 0 (m, 1 H) ,
7. 8 3 (d, 2 H, J = 8 H z ) ,
7. 9 - 7. 9 5 (m, 1 H) 実施例 2
2 - [4 - [3— 「 2— _( 4—クロ口フエニル) 5—プロピル— 4—ォキサゾ リル, プロピオニル]—フエニルォキシ] _— 2一メチルプロピオン酸 実施例 1 と同様な方法で以下の中間体ならびに標題化合物を得た。
( 1 ) 4一 (3— ( 2— (4一クロロフヱニル) 一 5—プロピル— 4—ォキサゾ リル) プロピオニル) フエノール 収率 6 7 %
1 H-NMR ( CD C 40 0MH z)
δ :
0 9 9 ( t 3 H, J = 7 H z ) ,
1 6 5 - 1 8 0 (m, 2 H) ,
2 72 ( t 2 H, J = 7 H z ) ,
2 9 1 ( t 2 H, J = 7 H z )
3 2 8 ( t 2 H, J = 7 H z) ,
6 7 0 (b r s 3 1 H) ,
6 8 1 (d, 2 H3 J = 8 H z ) ,
7 3 9 (d, 2 H, J = 8 H z) ,
7 8 1 (d, 2 H, J = 8 H z ) ,
7. 90 (d, 2 H, J = 8 H z)
(2) 2 - [4 - [3— [ 2 - (4—クロロフヱニル) 一 5—プロピル一 4—ォ キサゾリル] プロピオニル] フエニルォキシ] — 2—メチルプロピオン酸 ェチ ル 収率 59 %
δ : '
98 ( t 3 3 H, J = 7H z) ,
2 1 (t , 3 H, J二 7 H z) ,
64 ( s, 6 H ) ,
70 - 1. 80 (m, 2 H) ,
68 (t , 2 H, J = 7 H z ) ,
9 1 (t , 2 H, J = 7 H z) ,
32 (t, 2 H, J = 7 H z) ,
2 1 (q, , 2 H, J = 7 H z ) ,
82 (d, 2 H, J = 9 H z) ,
39 (d, 2 H, J = 8 H z) ,
89 (d, 2 H, J = 8 H z ) ,
9 1 (d, 2 H, J = 9 H z)
(3) 2 - [4 - [3— [2— (4—クロロフヱニル) 一 5—プロピル一 4—ォ キサゾリル] プロピオニル] フエニルォキシ] 一 2—メチルプロピオン酸
白色結晶
収率 84%
NMR (CDC 400MHz)
δ :
0. 98 ( t , 3 H, J = 7 H z )
1, 68 ( s , 6 H) ,
1. 70 - 1. 80 (m, 2 H) ,
2. 70 ( t 3 2 H, J = 7 H z
2. 9 1 ( t, 2 H, J = 7 H z
3. 24 ( t , 2 H, J = 7 H z
6. 89 (d, 2 H, J二 9 H z
7. 39 ( d , 2 H, J = 8 H z
7. 89 (d, 2 H, J = 8 H z
7. 89 (d, 2 H, J = 9 H z 実施例 3
2 - [4一 [3— [2— (2—クロ口フエニル) 一 5—イソプロビル一 4—ォキ サゾリル] ( 1—ヒ ドロキシィミノ) プロピル] フエニルォキシ] 一 2—メチル プロピオン酸
(1) 2 - [4— [3— [2— (2—クロ口フエニル) 一 5—イソプロビル一 4 —ォキサゾリル] ( 1ーヒドロキシィ ミノ) プロピル] フヱニルォキシ] — 2— メチルプロピオン酸ェチル
2 - [4一 [3— [ 2 - (2—クロ口フエニル) 一 5—イソプロピル一 4—ォ キサゾリル] プロピオニル] フエニルォキシ] - 2—メチルプロピオン酸ェチル (60m :, 0. 124 mm o l) 、 ヒドロキシルァミン塩酸塩 ( 26 m g , 0 . 372 mmo 1 ) をピリジン (2mL) —ェ夕ノ一ル ( 3 m L ) 中、 20時間 室温にて攙拌した。 反応物を水に注ぎ、 酢酸ェチルにて抽出した。 有機層を分取 後、 水、 食塩水で洗浄し無水硫酸ナトリウムで乾燥、 濾取した。 酢酸ェチルを減
圧留去し残渣に無色油状物である標題化合物 (45mg) を得た n (収率 74%)
1 H-NMR (CDC 133 400 MHz)
δ:
1. 20 ( t , 3H, J = 7 Η ζ ) ,
1. 22 (d, 6Η, J= 7Η ζ) ,
1. 58 (s , 6 Η) ,
2. 86 ( t , 2Η, J = 7 Η ζ ) ,
2. 95— 3. 05 (m, 1 Η) ,
3. 13 (t, 2 Η, J = 7 Η ζ ) ,
4. 19 (q, 2Η, J= 7Η ζ) ,
6. 78 ( d , 2Η, J = 9 Η ζ ) ,
7. 30 - 7. 40 (m, 2 Η) ,
7. 40 - 7. 50 (m, 1 Η) ,
7. 53 (d, 2Η, J = 9 Η ζ ) ,
7. 95— 8. 00 (m, 1 Η)
(2) 2— [4— [3— [2— (2—クロロフヱニル) 一5—イソプロピル一 4 一才キサゾリル] ( 1ーヒ ドロキシィ ミノ) プロピル] フエニルォキシ] —2— メチルプロピオン酸 上記のォキシム誘導体 (40 mg, 0. 08 mmo 1 ) をエタノール—水 ( 2 m L一 lmL) の混合溶媒に溶解させた後、 水酸化リチウム一水和物 ( 1 Omg) を加え、 室温にて 20時間攪拌した。 反応溶液を氷冷後、 希塩酸を加え中和した 後、 酢酸ェチルを加えた。 有機層を分取した後、 水で洗浄し無水硫酸ナトリウム で乾燥し濾取した。 酢酸ェチルを減圧留去し残渣に微黄色油状物である標題化合 物を 35 mg得た。
(収率 92%)
1 H-NMR (CD C 135 400 MHz)
0":
1. 22 (d, 6H, J = 7 H z ) ,
1. 60 ( s , 6 H) ,
2. 88 ( t , 2H, J = 7 H z ) ,
2. 95 - 3. 05 (m, 1 H) ,
3. 14 (t, 2 H, J = 7 H z ) ,
6. 82 (d, 2 H3 J = 9 H z ) ,
7. 25— 7. 40 (m, 2H) ,
7. 1 (d, 2 H, J = 9Hz)
7. 45— 7. 50 (m, 1 H) ,
7. 85— 7. 90 (m, 1 H) 実施例 4
2— 「4 [3 - [2——(2—クロ口フエニル 5ーィソプロピル—— 4ーォキ _ サゾリル] — 1— _ヒドロキシプロピル] フエニルォキシ] —2—メチルプロピオ ン睡
( 1) 4— [3— [2— (2—クロロフヱニル) 一 5—イソプロピル一 4ーォキ サゾリル] — 1—ヒ ドロキシプロピル] フエノール 水素化リチウムアルミニウム (25mg, 0. 659 mmo l) を THF (5 mL) に懸濁させた後、 氷冷下にて 4— [3— [2— (2—クロ口フエニル) 一 5ーィソプロピル一 4—ォキサゾリル] プロピオニル] フエノ一ル ( 240 mg , 0. 65mmo 1) を 3分間で添加した。 氷冷下で 1時間攪^した後、 更に室 温で 1時間攪拌した。 氷冷下にて飽和の塩化アンモニゥム水溶液を加えた後、 セ ライ ト濾過した。 水および酢酸ェチルを加え有機層を分取し、 有機層を水で洗浄 、 無水硫酸ナトリウムで乾燥し濾取した。 酢酸ェチルを減圧留去することで残渣 に微黄色油状物である標題化合物を 22 Omg得た。 (収率 9 1%)
1 H-NMR ( CD C 13 , 400 MHz)
δ :
1. 30 (d, 3H, J = 7 H z ) ,
1. 31 (d, 3H, J = 7 H z) ,
2. 0 - 2. 1 (m, 2 H) ,
2. 6 - 2. 8 (m, 2 H) ,
3. 00 - 3. 15 (m, 1 H3 ) ,
3. 9 - 4. 0 (b r , 1 H) ,
4. 77 (t, 1 H, J = 6Hz) ,
5. 6— 5. 7 (br, 1 H) ,
6. 76 ( 2 H, d, J = 8Hz) ,
7. 23 ( 2 H, d, J = 8Hz) ,
7. 30 - 7. 40 (m, 2 H) ,
7. 45 - 7. 50 (m, 1 H) ,
7. 95-8. 0 (m, 1 H)
(2) 2— [4一 [3— [2— (2—クロロフヱニル) 一 5—イソプロピル一 4 ーォキサゾリル] 一 1—ヒドロキシプロピル] フエニルォキシ] ー2—メチルプ 口ピオン酸 上記のフエノール化合物 (1 10mg, 0. 296 mmo 1)、 2—ブロモ— 2—メチルプロピオン酸ェチル ( 173mg, 0. 887 mmo 1) 、 炭酸力リ ゥム ( 122m :, 0. 887 mmo 1 ) をメチルェチルケトン (3mL) に懸 濁させた後、 20時間加熱還流した。 室温に戻した後、 不溶物を濾過、 更にメチ ルェチルケトンにて洗浄し溶媒を留去した。 この残渣をシリ力ゲルカラムクロマ トグラフィ一 (へキサン Z酢酸ェチル =5/1) で精製し 2— [4— [3— [2 ― (2—クロ口フエニル) 一 5—イソプロピル一 4—ォキサゾリル] 一 1—ヒ ド ロキシプロピル] フエニルォキシ] —2—メチルプロピオン酸 ェチルを無色油 状物 ( 125mg) として得た。 (収率 87 %)
このプロビンオン酸誘導体 ( 80 mg, 0. 1 6 mmo 1 ) をエタノール一水 (2mL- lmL) の混合溶媒に溶解させた後、 水酸化リチウ 水和物 ( 10 mg) を加えた。 室温にて 20時間攪拌後、 反応溶液を水冷し、 希塩酸を加え中 和、 酢酸ェチルを加え抽出した。 有機層を分取後、 水、 食塩水で洗浄し無水硫酸 ナトリウムで乾燥、 濾取した。 酢酸ェチルを減圧留去することで残渣に微黄色油 状物である標題化合物を 55mg得た。
(収率 73%)
1 H-NMR (CD C 133 400 MHz)
ό-:
1. 29 (d, 3 H3 J = 7 H z) ,
1. 31 (d, 3H, J二 7Hz) ,
1. 54 (s, 6H) ,
2. 0 - 2. 2 (m, 2 H) ,
2. 69 (t, 2 H, J = 7 H z ) ,
2. 95— 3. 15 (m, 1 H) ,
4. 78 (t, 1 H, J = 7 H z ) ,
6. 86 (d, 2H, J = 8 H z ) ,
7. 25 (d , 2 H, J = 8Hz) ,
7. 3 - 7. 4 (m, 2 H) ,
7. 45 - 7. 50 (m, 1 H) ,
7. 90 - 7. 95 (m, 1 H) 実施例 5
2— [ 4 [3— 「2——(2—クロ口フエ二ル)—一 5—イソプロピル一 4—ォキ サゾリル] 一— 1一プロぺニル 1 フエニルォキシ] — 2—メチルプロピオン酸
( 1) 4一 [3— [2— (2—クロロフヱニル) 一 5—イソプロピル一 4ーォキ サゾリル Ί プロぺニル] フエノール
4 - [3— [2— ( 2—クロ口フエニル) 一 5—イソプロピル— 4ーォキサゾ リル] 一 1ーヒドロキシプロピル] フエノール ( 1 1 0mg, 0 2 9 6 mmo 1) , パラ トルエンスルホン酸一水和物 ( 1 7mg, 0. 0 9 2 mmo 1 ) をト ルェン ( 2 mL) 中で 2 0時間加熱還流した。 原料の消失を確認後、 水にあけ 酢酸ェチルで抽出した。 有機層を分取後、 水で洗浄し無水硫酸ナトリウムで乾燥 し濾取した。 酢酸ェチルを減圧留去後、 残渣をシリカゲルカラムクロマトグラフ ィ一 (へキサン/酢酸ェチル = 5ノ1 ) で精製し上記の標題化合物を白色結晶 ( 3 Omg) として得た。
(収率 2 8 %)
1 H-NMR ( CD C 133 40 0 MH z)
ό· :
1. 34 (d, 6 H, J = 1 ζ) ,
3. 1 0— 3. 2 0 (m, 1 H) ,
3. 4 5 (d, 2 H, J = 6 H z) ,
6. 1 5 (d t , 1 H, J = 6 , 1 6 H z ) ,
6. 34 (d, 1 H, J = 1 6 H z ) ,
6. 3 6 ( s, 1 H) ,
6. 7 0 (d, 2 H, J = 8 H z ) ,
7. 0 9 (d, 2 H, J = 8 H z) ,
7. 3— 7. 4 (m, 2 H) ,
7. 4 - 7. 5 (m, 1 H) ,
7. 9 5 - 8. 0 0 (m, 1 H)
( 2 ) 2 - [4 - [3— [2— ( 2—クロロフヱニル) 一 5—イソプロピル一 4 一才キサゾリル] — 1—プロぺニル] フエニルォキシ] — 2—メチルプロビオン 酸ェチル
上述のフエノール誘導体 ( 3 0 mg, 0. 0 84mmo l ) 、 2—プロモー 2 一メチルプロビオン酸ェチル ( 5 0 mg, 0. 2 54 mmo 1 ) 、 炭酸カリウム (3 5 mg, 0. 2 54mmo 1 ) をメチルェチルケトン ( 3mL) に懸濁させ
た後、 2 0時間加熱還流した。 室温に戻した後、 不溶物を濾過、 専にメチルェチ ルケトンにて洗浄し溶媒を留去した。 この残渣をシリカゲルカラ クロマトグラ フィ一 (へキサン/酢酸ェチル = 5/ 1 ) で精製し上記の標題化合物を無色油状 物 ( 2 8 mg) として得た。
(収率 7 0 %)
H-NMR (CD C 135 40 0 MH z)
δ
24 (t , 3 H, J = 7 H z ) ,
3 2 (d, 6 H, J = 7 H z ) ,
54 ( s , 6 H) ,
3 0 5 - 3. 1 5 (m, 1 H) ,
3 4 7 (d d, 2 H, J = l , 6 H z) ,
4 2 2 (q 3 2 H, J = 7 H z ) ,
6 1 5 (d t , 1 H, J = 6, 1 6 H z ) ,
6 34 (d d, 1 E, J = X , 1 6 H z ) ,
6 7 7 (d, 2 H, J = 8 H z) ,
7 2 3 (d, 2 H, J = 8 H z) ,
7 3 0— 7. 40 (m, 2 H) ,
7 4 5 - 7. 5 0 (m, 1 H) ,
7 9 5 - 8. 00 (m5 1 H) .
( 3) 2— [4 - [3— [2— ( 2 -クロ口フエニル) 5—イソプロピル一 4 —ォキサゾリル] 一 1一プロぺニル] フエ二 2—メチルプロピオン 酸 上記のプロピオン酸誘導体 ( 2 5 mg, 0. 0 5 3 mmo 1 ) をェ夕ノ一ルー 水 ( 2 mL— l mL) の混合溶媒に溶解させた後、 水酸化リチウム一水和物 ( 6 mg) を加えた。 室温にて 2 0時間攪拌後、 反応溶液を氷冷、 希塩酸を加え中和 、 更に酢酸ェチルを加え抽出した。 有機層を分取後、 水、 食塩水で洗浄し無水硫
酸ナトリウムで乾燥、 濾取した。 酢酸ェチルを減圧留去し残渣に無色油状物であ る標題化合物を 1 5mg得た。 (収率 63 %)
1 H-NMR ( CD C 13 , 400 MHz)
δ :
1. 33 (d, 6H, J = 7Hz) ,
1. 57 ( s , 6 Η) ,
3. 05 - 3. 20 (m, 1 Η) ,
3. 48 (dd, 2Η, J = 1 , 6 Η ζ ) ,
6. 20 - 6. 30 (m, 1 Η) ,
6. 42 (dd, 1 Η, J= l, 1 6 Η ζ ) ,
6. 86 (d, 2 Η, J = 8 Η ζ) ,
7. 22 (d, 2Η, J = 8Hz) ,
7. 30 - 7. 40 (m, 2 Η) ,
7. 45 - 7. 50 (m, 1 Η) ,
7. 95— 8. 00 (m, 1 Η) . 実施例 6
2 - [ 4 - [3— [ 2 - (2—クロ口フエニル) 一 5—イソプロピル一 4ーォキ サゾリル] プロピル] フエニルォキシ] —2—メチルプロピオン酸
2— [4 - [3— [ 2 - (2—クロ口フエニル) 一 5—イソプロピル一 4一才 キサゾリル] — 1—プロピニル] フエニルォキシ] 一 2—メチルプロピオン酸 ェチル (40mg, 0. 085 mmo 1) のエタノール (8mL) 溶液に, 10 %P t -C (8mg) を加えた後、 水素雰囲気下 (常圧) にて 8時間攪拌した。 ォレフィン体の消失を確認した後、 水 (3mL) ならびに水酸化リチウム一水 和物 (6mg) を加え 20時間攪拌した。 反応溶液を氷冷し、 希塩酸を加え中和 後、 酢酸ェチルを加えた。 有機層を分取した後、 水で洗浄し無水硫酸ナトリウム で乾燥し濾取した。 酢酸ェチルを減圧留去し、 残渣をシリカゲルカラムクロマト グラフィー (へキサン/酢酸ェチル = 5/1) で精製し上記の標題化合物を無色
油状物 ( 1 7mg) として得た
( 2工程収率 4 5 %)
NMR ( CD C 133 4 0 0 MH z)
δ :
1. 2 9 (d, 6 H, J = l E z )
5 6 ( s 5 6 H) ,
1. 8 5 - 2. 0 5 (m 2 H) ,
2. 5 6 ( t , 2 H, J 7 H z )
2. 6 5 ( t , 2 H , J 7 H z )
2. 9 5 - 3. 1 0 (m 1 H) ,
6. 8 7 (d, 2 H, J 8 H z )
7. 1 1 (d3 2 H, J 8 H z )
7. 3 0 - 7. 40 (m 2 H) ,
7. 4 5 - 7. 50 (m 1 H) ,
7. 9 0 - 7. 9 5 (m 1 H) . 実施例 7
実施例 1記載の方法と同様な方法で以下の化合物を得た。
( 7 - 1 )
2 - [4— [2— ( 2—クロ口フエニル) 一 5—イソプロピル一 4ーォキサゾ リル] メチルカルバモイル] フエニルォキシ— 2 _メチルプロピオン酸 白色結晶
m p 1 2 0 - 1 2 1 °C
1 H-NMR (CD C I" 4 0 0 MH z)
δ;
1. 3 4 ( 6 H, d, J = 7 H z ) ,
1. 6 5 ( 6 H5 s) ,
3. 41 ( 1 H, q q , J = 7 H z 7Hz)
4. 53 ( 2 H, d, J = 6 Hz)
6. 89 (2H, d, J = 9 H z )
7. 2— 7. 4 ( 2 H, m) ,
7. 47 ( 1 H, d d , J = 1 H z 8 H z ) ,
7. 67 ( 1 H5 d d , J = 1 H z 8 H z ) ,
7. 69 ( 2 H, d, J = 9 Hz) ,
7. 79 ( 1 H5 t, J = 6 Hz) .
I R vm a x ( K B r ) cm"1 ;
3381, 3377, 2974, 1701, 1697 , 1 662, 1605, 1574, 1541 , 1500, 1460, 1439, 1385, 1288, 1246, 1 188, 1 1 55, 1053, 1022, 966, 9 10, 850, 796, 766, 737, 654, 636, 592.
(7-2)
2 - [4 [2——(2—クロロフェニル) — 5—イソプロピル一— 4—ォキサ リル] ァセチルァミノ Ί フエニルォキシ一 2—メチルプロピオン酸 白色アモルファス
H-NMR ( C D C 13 , 400 MHz)
δ;
1. 35 (6H, d, J = 7 Hz) ,
1. 56 (6H, s) ,
3. 25 ( 1 H, m) ,
3. 67 ( 2 H, s ) ,
6. 88 ( 2 H, d, J = 9 Hz) ,
7. 35〜 7. 40 ( 2E, m) ,
7. 43 ( 2 H, d, J = 9 H z ) ,
7. 53 ( 1 H, m) ,
7. 93 ( 1 H, m) ,
9. 39 ( 1 H, s ) .
(7-3)
2— [4一 [4— [2— (2—クロ口フエニル) 一 5—イソプロピル一 4ーォ キサゾリル] プチリル] フエニルォキシ] —2—メチルプロピオン酸 黄色油状物
1 H-NMR (CD C 13 400 MHz)
δ;
1. 30 (d, 6 H, J = 7Hz)
1. 66 ( s, 6 H)
2 · 04 (m, 2 H)
2. 62 ( t, 2 H, J二 7 H z )
2. 9 1 ( t , 2 H, J = 7 H z )
3. 10 (m, 1 H)
6. 9 1 (d, 2 H, J = 9 H z )
7. 3 - 7. 5 (m, 3 H)
7. 87 ( d , 2 H, J = 9 Hz)
7. 91 (m, 1 H)
(7-4)
2 - [4— C 3 - [2— (2, 4—ジクロ口フエニル) 一 5—イソプロピル一 4—ォキサゾリル] プロピオニル] フエニルォキシ] —2—メチルプロピオン酸 白色結晶 mp 100 - 105 °C
1 H-NMR (CDC 13, 400 MHz)
δ;
1. 30 (d, 6H, J = 7 H z ) ,
. 65 ( s , 6 H) ,
. 95 (t, 2 H3 J = 7Hz) ,
. 20 ( q q, 1H, J = 7 H z , J = 7 H z )
. 28 (t, 2 H, J = 7 H z ) ,
. 88 (d, 2 H5 J = 8 H z ) ,
. 29 (dd, 1 H3 J = 2. 9 H z ) ,
. 49 (d, 1 H, J = 2 H z ) ,
. 85 (d, 1 H, J = 9Hz)
. 85 (d, 2H, J = 8Hz)
(7- 5)
2— [4— [3— C 2 - (2, 4—ジクロ口フエニル) 一 5—イソプロピル 4ーォキサゾ'リル] プロピオニル] フエニルォキシ] —2—メチルプチリル酸 微黄色アモルファス
1 H-NM (CDC 1" 400 MHz)
δ;
1. 00 (t, 3 H, J = 7 H z ) ,
1. 30 (d, 6 H, J = 7 H z ) ,
1. 57 ( s, 3 H) ,
1. 90-2. 10 (m, 2 H) ,
2. 95 (t, 2H, J = 7 H z ) ,
3. 15-3. 40 (m, 3 H) ,
6. 90 (d, 2 H, J = 8 H z ) ,
7. 30 (dd, 1H, J = 2. 9 Hz)
7. 49 (d, 1 H, J = 2 H z ) ,
7. 88 (d, 1 H, J = 9 H z ) ,
7. 90 (d, 2 H3 J = 8 H z )
( 7 - 6)
2 - [4— [3— C 2 - ( 2 , 3—ジクロロフエニル) 一 5— ソプロピル一 4—ォキサゾリル] プロピオニル] フエニルォキシ] 一 2—メチルプロピオン酸 白色アモルファス
1 H-NMR (CD C 13, 40 0 MH z)
δ;
1. 3 1 (d, 6 H, J = 7 H z ) ,
1. 6 9 ( s , 6 H ) ,
2. 9 3 ( t , 2 H, J = 7 H z ) ,
3. 2〜 3. 3 (m, 3 H) ,
6. 8 9 (d, 2 H, J = 9 H z ) ,
7. 49 (d, 1 H, J = 8 H z ) ,
7. 7 8 (d d5 1 H, J = 2 and 8 H z ) ,
7. 8 1 (d, 2 H, J = 9.H z ) ,
8. 0 3 (d, 1 H, J = 2 H z ) .
( 7 - 7)
2— [4— 「3— [2— ( 2—ペンゾフラニル) 一 5—イソプロピル一 4—ォ キサゾリル] プロピオニル] フエニルォキシ] 一 2—メチルプロピオン睡俊 微褐色結晶
m 1 3 5 - 1 3 9 °C
1 H-NMR (CD C 133 4 0 0 MH z)
δ
1. 3 3 (d, 6 H, J = 7 H z)
1. 6 9 ( s , 6 H)
2. 9 6 ( t , 2 H, J = 7 H z )
3. 24 (m, 1 H)
3. 3 2 ( t, 2 H, J = 7 H z )
6. 9 1 (d, 2 H, J = 9 H z )
7. 2 - 7. 3 (m, 2 H)
7. 3 6 (m, 1 H)
7. 5 5 (d, 1 H, J = 8 H z )
7. 6 2 (d, 1 H, J = 8 H z )
7. 8 6 (d, 2 H, J = 9 H z )
I R レ max ( K B r ) cm"1 ;
2 9 6 8 , 1 7 1 3 , 1 6 8 0 , 1 6 3 3 , 1 5 9 9 , 1 5 7 2 1 5 04 1 4 7 0, 1 4 1 2 , 1 3 6 0, 1 3 0 2, 1 2 5 7 , 1 2 1 5 1 149 1 1 1 1 , 1 0 3 2 , 9 64 , 84 9 , 8 1 6 , 7 44.
( 7 - 8)
2 - [4 [ 3— 「 2——( 4—ビフエ二リル) 一—5—イソプロピル一 4—ォキ サゾリル] プロピオニル Ί_フェニルォキシ]—一 2—メチルプロピオン酉変_ 淡黄色アモルファス
1 H-NMR (CD C 13 , 40 0 MH z )
δ;
1. 3 2 (d, 6 H, J = 7 H z )
1. 6 9 ( s, 6 H)
2. 9 5 ( t, 2 H, J = 7 H z )
3. 1 - 3. 3 (m, 3 H)
6. 8 9 (d, 2 H, J = 9 H z )
7. 3 - 7. 7 (m, 7 H)
7. 7 7 (d, 2 H, J = 9 H z )
8. 0 2 (d, 2 H5 J = 8 H z )
( 7 - 9 )
2 - [4— [ 3— [ 2— ( 1 —ヒ ドロキシ一 2—ナフチル) ― 一イソプロピ ルー 4—ォキサゾリル] プロピオニル] —フエニルォキシ] 一 2—メチルプロピ オン酸 淡黄色アモルファス
1 H-NMR (C D C 13 4 0 0 MH z )
3 0 d , 6 H, J = 7 H z ) ,
6 6 s , 6 H) ,
2 9 8 t , 2 H, J = 7 H z ) ,
3 2 2 q q, 1 H, J二 7 H z , 7 H z )
3 3 6 t , 2 H, J = 7 H z ) ,
6 9 2 d, 2 H, J = 8 H z ) ,
7 3 7 d, 1 H, J = 9 H z )
7 4一 7 . 6 (m, 2 H) ,
7 7 7 d d , 1 H5 J = 2 , 9 H z ) ,
7 8 1 d , 1 H, J = 9 H z )
7 9 4 d, 2 H, J = 8 H z )
8 3 9 d d, 1 H, J = 2 , 9 H z )
( 7 - 1 0 )
2 - [ 4 - [ 3— [ 2— ( 2 , 4—ジクロロフエニル) 5—イソプロピル一
4—ォキサゾリル 1一 1—プロぺニル 1 フェニルォキシ 1 - 2一メチルプロピオ ン酸 淡黄色油状物
1 H-NMR (CD C 1" 4 0 0 MH z )
δ:
1. 33 (d, 6 H, J = 7 H z ) ,
1. 57 ( s, 6 H) ,
3. 10 (m, 1 H) ,
3. 48 (dd, 2 H, J = l, 6Hz) ,
6. 25 (m, 1 H) ,
6. 42 (dd, 1 H, J = 13 16 H z ) ,
6. 86 (d, 2 H, J = 8 H z ) ,
7. 25 - 7. 35 (m, 2 H)
7. 50 (d, 1 H, J = 2Hz)
7. 92 (d, 2 H, J = 8 H z )
(7- 1 1)
2— [4 - [3— [ 2 - (3—ヒドロキシ一 2—ナフチル) 一 5—イソプロピ ルー 4—ォキサゾリル 1プロピオニル] フエニルォキシ 1 — 2—メチルプロピオ ン酉
黄色アモルファス
1 H-NMR (CD C 13 400 MHz)
δ;
1. 37 (d 6 H, J 7 H z ) 5
1. 66 ( s 6 H)
97 ( t 2 H, J 7 H z ) ,
25 (m 1 H)
35 ( t 2 H, J 7 H z ) ,
92 (d 2 H, J 9 H z ) ,
30 (m 1 H)
35 ( s 1 H)
43 (m 1 H)
68 (d 1 H5 J = 7 H z )
7. 8 1 (d, 1 H, J 7 H z )
7. 9 3 (d, 2 H, J 9 H z )
8. 3 0 ( s , 1 H) .
( 7 - 1 2 )
2 - [4— [ 3— [ 2— (4—クロ口一 2—ヒ ドロキシフエニル) 一 5—イソ プロピル一 4—ォキサゾリル] プロピオニル] フエニルォキシ] 一 2—メチルプ 口ピオン酸 白色アモルファス
H-NMR ( C D C 1 40 0 MH z)
δ;
1 3 1 d 6 Η, J = 7 Η ζ ) ,
1 6 7 s 6 Η) ,
2 9 4 t 2 Η, J = 7 Η ζ ) ,
3 2 0 m 1 Η) ,
3 3 1 ΐ 2 Η, J二 7 Η ζ ) ,
6 9 0 d d , 1 Η, J二 2 and 9 H z ) ,
6 9 3 d, 2 H, J = 9 H z ) ,
7 0 3 d, 1 H, J = 2 H z ) ,
7 6 8 d, 1 H, J = 9 H z ) ,
7 9 1 d, 2 H, J = 9 H z ) .
( 7 - 1 3 )
2— [4— [3— 「 2— (4—プロモー 2—クロ口フエニル) 一 5—イソプロ ピル一 4—ォキサゾリル] プロピオニル] フエニルォキシ] — 2—メチルプロピ オン酸 白色アモルファス
1 H-NMR (CD C 135 40 0 MH z)
δ;
3 0 (d, 6 H, J = 7 H z )
6 5 ( s , 6 H)
9 5 ( t , 2 H, J = 7 H z )
2 0 (m, 1 H)
2 6 ( t , 2 E, J二 7 H z )
8 7 (d, 2 H, J = 9 H z )
43 ( d d, 1 H, J = 2, 8 H z )
6 5 ( d, 1 H, J = 2 H z )
7 6 ( d, 1 H, J = 8 H z )
8 3 ( d, 2 H, J = 9 H z )
( 7 - 1 )
2— [4— [3— [2— ( 3—クロ口一 4ービフエ二. 5—イソプロビ ルー 4—ォキサゾリル 1 プロピオニル 1 フエニルォキシ] 一 2—メチルプロピオ ン歴 白色ァモルファス
1 H-NMR (CD C 133 40 0 MH z)
δ;
1. 3 2 ( d, 6 H, J二 7 H z )
1. 6 5 ( s , 6 H)
2. 9 7 ( t , 2 H, J = 7 H z )
3. 2 2 (m, 1 H)
3. 2 7 ( t , 2 H, J = 7 H z )
6. 8 8 (d, 2 H, J二 9 H z )
7. 3 - 7. 5 (m, 3 H)
7. 5 3 ( d d, 1 H, J = 2, 8 H z)
7. 5 - 7. 6 (m, 2 H)
7. 71 (d, 1 H, J = 2 H z )
7. 82 (d, 1 H, J = 9 H z )
7. 95 (d, 2H, J = 8 H z ) 実施例 8
(8- 1)
実施例 1記載の方法と同様な方法で以下の化合物を得た。
2 - [4— [3— [2— [ ( 4— トリフルォロメチル) フエニル] —4一メチル —5—チアゾリル] プロピオニル] フエニルォキシ] —2—メチルプロピオン酸 白色結晶 mp 158 - 160 °C
XH-NMR ( CD C 1 a, 400 MHz)
δ;
1. 68 ( s , 6 H) ,
2. 45 ( s, 3 H) ,
3. 2 - 3. 35 (m, 4 H)
6. 93 (d, 2H3 J = 9Hz) ,
7. 63 (d, 2H, J二 9Hz) ,
7. 9 1 (d, 2H, J = 9 H z ) ,
7. 96 (d, 2H, J = 9 H z ) .
(8-2)
実施例 1記載の方法と同様な方法で以下の化合物を得た。
2 - [3——「3——[2— ( 2 ,—4—ジクロ口フエニル) 一 5—イソプロピル一 4 ーォキサゾリル] プロピオニル] フエニルォキシ 1 一 2—メチルプロピオン睡 白色結晶 mp 1 15 - 120 °C
^-NMR ( CD C 133 400 MHz)
δ;
1. 31 (d, 6H, J = 7 H z ) ,
1. 6 2 ( s, 6 H) ,
2. 9 2 (t , 2 H, J = 7 H z ) ,
3. 1 6 (q q , 1 H, J = 7 H z , J = 7 H z ) ,
3. 24 (t, 2 H, J = 7 H z ) ,
7. 1 6 (d d, 1 H, J = 2 , 9 H z ) ,
7. 3 4 (d d, 1 H, J = 2 , 9 H z ) ,
7. 3 5 (t , 1 H, J = 9 H z ) ,
7. 5 1 (d, 1 H3 J = 2 H z ) ,
7. 6 - 7. 7 (m, 2 H) ,
7. 8 8 (d, 1 H, J = 9 H z )
( 8 - 3)
2— [4— [3— C 2 - ( 2 , 4—ジクロロフエニル) 一 5—イソプロピル一 4 ーォキサゾリル Ί プロピオニル]フエニルスルファニル Ί 一 2一メチルプロピオ ン i
( 1 ) 3— [2— ( 2 , 4—ジクロロフエニル) 一 5—イソプロピル一 4ーォキ サゾリル] — 1— ( 4—ヒドロキシフエニル) プロパン一 1—オン
氷冷した THF ( 1 5 mL) に 6 0 %水素化ナトリウム ( 1 2 0mg, 3. 0 0 mm o 1 ) を加えた。 続いて 2 - [ ( 4一ペンジルォキシ) ベンゾィル] 酢酸 ェチル ( 9 0 0mg, 3. 0 2 mmo 1) の T H F ( 1 5 m L ) 溶液を 3 0分間 で滴下した。 室温に戻し 3 0分攪拌した後、 4—ョ一ドメチルー 5—イソプロ'ピ ルー 2— ( 2 , 4—ジクロ口フエニル) ォキサゾ一ル ( 1. 2 0 g, 3 · 0 0m mo 1 ) を加えた。 窒素雰囲気下にて 2 0時間加熱還流した後、 室温に戻し T H Fを減圧留去した。 残渣に酢酸 ( 7. 5mL) —濃塩酸 ( 2. OmL) を加え 5 時間加熱還流後、 室温に戻し氷冷水にあけ、 酢酸ェチルを加え有機層を分取した 。 有機層を分取後、 飽和の炭酸水素ナトリウム水溶液、 水、 食塩水で洗浄し無水 硫酸ナトリウムで乾燥、 濾取した。 酢酸ェチルを減圧留去後、 残渣をシリカゲル カラムクロマトグラフィー (へキサン/酢酸ェチル = 3/ 1 ) にて精製し上記の
標題化合物を微黄白色結晶 ( 6 5 Omg) として得た。 (収率 5 3 %)
^-NMR ( CD C 133 4 0 0 MH z)
δ :
1. 3 2 (d, 6 H, J = 7 H z ) ,
9 6 ( t , 2 H, J = 7 H z ) ,
2 2 (q q, 1 H, J = 7 H z, 7 H z ) ,
2 5 ( t, 2 H, J = 7 H z ) ,
7 7 (d, 2 H, J = 8 H z ) ,
2 9 (d d, 1 H, J = 2 , 8 H z ) ,
4 9 (d, 1 H5 J = 8 H z ) ,
6 0 (b r s , 1 H) ,
7 6 (d, 2 H, J = 8 H z ) ,
84 (d, 1 H, J = 8 H z ) ,
(2) 3 - [2— ( 2 , 4—ジクロ口フエニル) 一 5—イソプロピル一 4—ォキ サゾリル] — 1— [ ( 4一ジメチルチオ力ルバモイルォキシ) フエニル] プロパ ン一 1一オン
上記のフエノール誘導体 ( 1. 0 0 g, 2. 4 7mmo l ) 、 4—ジメチルァ ミノピリジン (3 0mg, 0 · 2 5 mmo 1 ) ヽ トリェチルァミン ( 0. 7 mL , 4. 9 mmo 1 ) を d r yジォキサン ( 5. 0 mL) に溶解させた後、 氷冷 下にて塩化ジメチルチオ力ルバモイル ( 3 6 7 mg, 2. 9 7 mmo 1 ) を加え た。 反応温度を上げ一晩加熱還流した後、 室温に戻し氷冷水にあけ、 酢酸ェチル を加え有機層を分取した。 有機層を飽和の炭酸水素ナトリウム水溶液、 水、 食塩 水で洗浄後、 無水硫酸ナトリウムで乾燥、 濾取した。 酢酸ェチルを減圧留去後、 残渣をシリカゲルカラムクロマトグラフィー (へキサン/酢酸ェチル = 3/ 1 ) にて精製することで上記の標題化合物を微黄色油状物 ( 1. 1 5 g) として得た 。 (収率 9 5 %)
^-NMR (CD C 13, 40 0 MH z)
o
丄 · 丄 ^ Q! , R Ό X JUL J J T ί xl Z a
Q Q
Δ · 、 , Δ J J ί -tl Z 3
O, —
ά U ( q q 3 1 H, J —— ί X 1T1 Z 7 ( ti Z )
0 0 s, o ) 3
q Q
, 、 rl J ί7 T -tJi Zヽ 3
V s y 0 i ) 3
. 丄 ΰ TJ T o 1 Z ,
O n U 丄 J , J ― Λ
7. 49 (d, 1 H, J 2 H z ) ,
7. 89 (d, 1 H, J 9 H z ) 3
8. 04 (d , 2 H, J 8 H z ) ,
( 3 ) 3 - [2— (2, 4—一ジクロロフエニル) 一 5—イソプロピル一 4—ォ キサゾリル] — 1— [ ( 4—ジメチルカルバモイルスルファニル) フエニル] プ 口パン一 1 _オン
上記のチォカルバモイル体 ( 1. 10 g, 2. 24 mmo 1 ) を n—テトラデ カン ( 15mL) に溶解させた後、 内温 250°Cにて 8時間加熱還流した。 室温 に戻した後、 反応溶液を直接、 シリカゲルカラムクロマトグラフィー (へキサン /酢酸ェチル二 3Z1 ) に付し精製することで上記の標題化合物を微黄色油状物 (35 Omg) として得た。 (収率 3 1%)
δ :
1. 31 (d, 6H, J = 7 H z )
2. 98 (t, 2 H, J = 7 H z ) ,
3. 0— 3. 2 (b r, 6 H) ,
3. 19 ( q q 3 1 H, J = 7 H z , 7 H z ) ,
3. 39 ( t , 2 H, J = 7 H z ) ,
7. 30 (dd, 1 H, J = 2, 9 H z) ,
7. 49 (d, 1 H, J = 2 H z )
7. 58 (d, 2H, J = 8Hz)
7. 88 (d, 1 H, J = 9Hz)
7. 98 (d, 2H, J = 8Hz)
(4) 3 - [2— (2, 4—ジクロロフエニル) 一 5—イソプロピル一 4—ォキ サゾリル] ー 1一 ( 4—メルカプトフエニル) プロパン一 1一オン
上記の力ルバモイル体 (335mg, 0. 68 mm o 1 ) を dryメ夕ノ一ル
(8mL) に溶解させた後、 0. 5N Me ON a ( 2. OmL) を加え 20時 間加熱還流した。 室温に戻した後、 反応溶液を氷冷水にあけ、 3 N塩酸水溶液を 加え中和後、 酢酸ェチルを加え有機層を分取した。 有機層を水、 食塩水で洗浄し 無水硫酸ナトリウムで乾燥、 濾取した後、 酢酸ェチルを減圧留去することで、 残 渣に粗体の標題化合物を微黄白色固体 (277 mg) として得た。 (粗収率 97 %)
U MR ( CD C 133 400 MHz)
δ :
1. 30 (d, 6 H, J = 7Hz) ,
2. 96 (t, 2 H, J = 7Hz) ,
3. 16 ( q q, 1 H, J二 7Hz, 7 H z ) ,
3. 24 (t, 2H, J = 7Hz) ,
3. 60 ( s, 1 H) ,
7. 2— 7. 3 (m, 3 H) ,
7. 49 (d, 1 H, J = 2Hz) ,
7. 84 (d, 2 H, J = 8Hz) ,
7. 87 (d, 1 H, J = 9 Hz) ,
( 5 ) 2 - [4— [3— 「2— (2, 4—ジクロロフエニル) 一5—イソプロピ ルー 4—ォキサゾリル 1 プロピオニル] フエニルスルファニル] 一 2—メチルプ 口ピオン酸
上記 (4 ) で得られた 3— [ 2— ( 2, 4ージクロ口フエニル) 一 5—イソプ 口ビル一 4—ォキサゾリル] — 1 — ( 4一メルカプトフエニル) 口パン一 1 一 オンを用い、 実施例 1の (2 ) 、 ( 3 ) と同様な方法により標記化合物を得た。 微黄色アモルファス
一 NMR ( CD C 13 4 0 0 MH z )
δ;
1. 3 0 ( d , 6 H, J : 7 H z )
1. 5 2 ( s, 6 H) ,
9 7 ( t, 2 H, J = 7 H z ) ,
1 9 ( q q , 1 H, J = 7 H z , 7 H z ) ,
3 7 ( t , 2 H, J二 7 H z ) ,
2 9 ( d d, 1 H, J = 2 , 8 H z ) ,
4 8 ( d , 1 H, J = 2 H z ) ,
5 5 ( d , 2 H, J = 9 H z )
8 6 ( d , 1 H, J = 8 H z ) ,
9 0 ( d, 2 H, J = 9 H z )
( 8 - 4 )
実施例 1記載の方法と同様な方法で以下の化合物を得た。
2— [ 4— 13 - [ 2— [ ( 4一 トリフルォロメチル) _フヱニル] 4—イソプ 口ピル一 5—チアゾリル] プロピオニル]—フエニルォキシ] 一 2—メチルプロピ オン涯
白色アモルファス
一 NMR ( C D C 135 4 0 0 MH z )
δ;
1. 3 3 ( d , 6 H, J = 7 H z ) ,
1. 6 8 ( s , 6 H) ,
3. 1 5 ( q q, 1 H, J = 7 H z 5 J = 7 H z ) ,
3. 2 - 3. 3 (m, 4 H) ,
6. 94 (d, 2H, J = 9Hz) ,
7. 64 (d, 2H, J = 8 H z ) ,
7. 92 (d, 2H, J = 9 Hz) ,
7. 99 (d, 2 H3 J = 8Hz) , 実施例 9
(薬理実験)
I.測定方法
(l)PPARa, Ί、 δ活性化能の測定
試験化合物 〔実施例 1〜 6及び既知の P PAR 5ァゴニストの L一 16504 1 (B e r g e r , J. 他, ( 1999) J. B i o l. C h e m. , 274 : 6718- 6725) 〕 の PPARひ、 ァ及び d活性化能を以下のように測定し た。
1) 材料
アフ リカミ ドリザル腎線維芽細胞 (( ¥— 1細胞) は, 東北大学加齢医学研究 所 医用細胞資源セン夕一より入手した。 すべての試験化合物は, ジメチルスル ホキシド (DMSO) に溶解し, 最終 DMSO濃度 0. 1%で試験に用いた。
2) プラスミ ド
受容体発現プラスミ ド (GAL 4— hPPARひ LBD、 GAL 4 -hP P ARァ LBD、 GAL4— ; hPPARS LBD) , レポ一夕一プラスミ ド ( UASx4-TK-LUC) , ?—ガラク トシダ一ゼ発現プラスミ ド ( G A L ) は K l i ewe r, S . A. 他, ( (1992) Nat ur e, 358 : 77 1- 77 ) と同様のものを使用した。
3 ) トランスフエクシヨン
C V- 1細胞を 1ゥエル当たり 2 X 105個の細胞濃度で, 24ゥエル培養プ レートに播き, 24時間, 4 %胎児ゥシ血清 (F C S) 添加 OP T I— MEM I Reduc e d S e rum Med ium (L i f e T e chno l o
g i Θ s ) 500 1/we 11で培養した。 その後, 血清無添加の OP T I— MEMで細胞を洗い, DNA含有溶液 〔 1ゥヱル ( 250〃 1 溶液) 当たり , 以下の成分を含有するもの ; 0. 03〃gの GAL4— hPPAR<5 L B D , 0. 25 /gの UASx4— TK— LUC, 0. 35 zgの/? GAL、 2 μ 1 のリポフエクション試薬 D MR I Ε - C (L i f e Te chno l o g i e s ) , これらを OP Τ I—MEMに溶解し, 室温で 30分間静置したもの〕 を添加 して, 37°Cで 5時間培養した。
4) 試験化合物添加による細胞処理
DNA含有溶液を除き, 試験化合物 (終濃度 : 10— 4Mあるいは' 10— 5Μにな るように 100 %DM S 0に溶解したもの) を含む 4 %F CS-0PT I-ME M 500〃1に新たに交換してさらに 40時間, 37°Cで培養した。
5) レポーター遺伝子発現レベルの測定
培地を除き, PB Sで 2回洗った後, 凍結融解を 1回行い, 1ゥエル当たり, ルシフヱラ一ゼ活性測定用可溶化緩衝液 (25mM T r i s -P04 (p H 7. 8) , 15 % v/v Gl ys e ro l, 2 % C H AP S , 1 % L e c i t h i n, 1 %B S A, 4mM E GT A (p H 8. 0) , 8 mM MgC l2 , ImM DTT) 100 z 1を添加して, 室温で 10分間放置した。 そのうち の 20〃 1を 96ゥエル測定用プレートに分取して, ルシフェラーゼ基質溶液 1 00〃1 (ピツカジーン ; 二ヅポンジーン社製) を添加し, MLR— 100型マ ィクロルミノリーダ (コロナ電気社製) を用いて, 1秒間の発光量 (ルシフェラ —ゼ活性) を求めた。 ルシフェラ一ゼ遺伝子の添加と同時に加えておいた/? GA Lの細胞内導入による活性発現量を測定し, 化合物添加によるルシフェラーゼ活 性の変動を導入遺伝子のトランスフヱクシヨン効率で補正した。 ーガラク トシ ダ一ゼ活性の測定方法は, 50〃 1の可溶化試料を別な 96ゥエルプレートに分 取し, ONPG (2—二トロフエニル一^— D—ガラク トビラノシド) 溶液 10 0〃1を添加して, 室温で 5分間インキュベートした。 反応停止液 ( 1M炭酸ナ トリウム溶液) 50〃1を加え, 414 nmの吸光度を測定した。 溶媒として用 いた DMS0 (0. 1%濃度) のみで処理した細胞のルシフヱラ一ゼ活性値 (コ ントロ一ル値) を 0 %に, 対照薬 (PPARひ : 1 (T4M WY- 165041
, PP ARr: 10"5M Ro s i g l i t az one, PPARd 10- 4M L— 16504 1 ) で処理した細胞のルシフェラ一ゼ活性値を 00 %として , 相対的な PPAR活性化能を算出した。 試験結果
試験結果表 25に示す ( 【表 25】
P PAR活性化能:対照薬を 100%としたときの相対値
PPARひ、 ァ、 d活性化能の測定は試験化合物濃度が 10— 5Mの値 表 25から明らかなように、 実施例化合物は L— 1 65041と比べ、 同等又 はそれ以上の強力な P P A R (5活性化能を有することが明らかになった。 実施例 10
実施例 7記載の化合物に関し、 実施例 9と同様な試験方法で P PAR活性化能 を測定した。 その結果を表 26に示す。
【表 2 6】
表 2 6から明らかなように、 本発明化合物 (実施例 7— 4, 実施例 7— 6 , 実 施例 7— 1 1等) は L一 1 6 5◦ 41と比べ、 同等又はそれ以上の強力な P P A 活性化能を有することが明らかになった。 また本発明化合物 (実施例 7— 4 , 実施例 7— 1 2等) については、 P PARひ及び y活性化能に比べ、 PPAR δに対し選択性の高い活性化能を示した。 実施例 1 1
実施例 8記載の化合物に関し、 実施例 9と同様な試験方法で PPAR活性化能 を測定した。 その結果を表 2 7に示す。
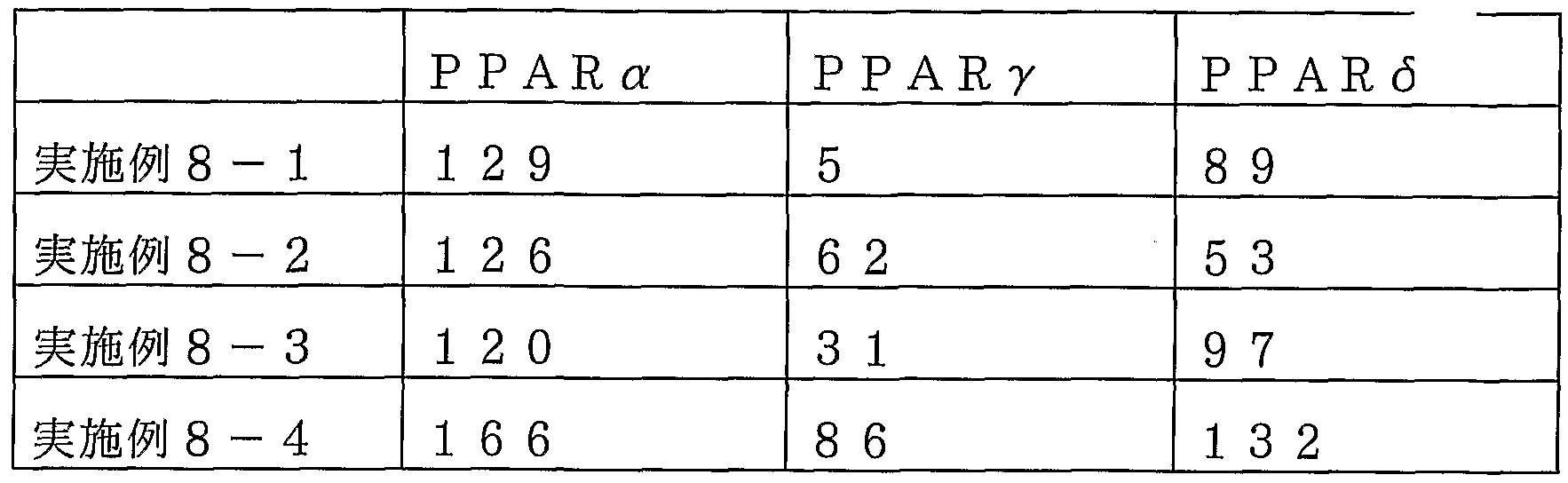
【表 27】
PPAR活性化能:対照薬を 100 %とした時の試験化合物 10_5Mでの 相対値
PPARa : WY- 14643 ( 1 0~4M)
PPARァ : Ro s i gl i t a z one ( 10一5 M)
PPAR(5 : L- 165041 ( 1 0~4M) 表 27から明らかなように、 本発明化合物 (実施例 8— 1〜実施例 8— 4) は L- 165041と比べ、 同等又はそれ以上の強力な P PAR d活性化能を有す ることが明らかになった。