WO2007059871A1 - Hydroxy-substituierte diphenylazetidinone zur behandlung von hyperlipidämie - Google Patents
Hydroxy-substituierte diphenylazetidinone zur behandlung von hyperlipidämie Download PDFInfo
- Publication number
- WO2007059871A1 WO2007059871A1 PCT/EP2006/010840 EP2006010840W WO2007059871A1 WO 2007059871 A1 WO2007059871 A1 WO 2007059871A1 EP 2006010840 W EP2006010840 W EP 2006010840W WO 2007059871 A1 WO2007059871 A1 WO 2007059871A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- phenyl
- inhibitors
- radical
- substituted
- Prior art date
Links
- 0 *N(CC1)CCN1C(Oc1cc(O)c([C@@]([C@@](CCC(c(cc2)ccc2F)O)C2=O)N2c(cc2)ccc2F)cc1)=O Chemical compound *N(CC1)CCN1C(Oc1cc(O)c([C@@]([C@@](CCC(c(cc2)ccc2F)O)C2=O)N2c(cc2)ccc2F)cc1)=O 0.000 description 1
- COMSZISWKMWFBA-VYOXFEDNSA-N CCN1CCN(Cc2ccc(COc3ccc([C@H](C(CC[C@@H](c(cc4)ccc4F)O)C4=O)N4c(cc4)ccc4F)c(O)c3)cc2)CC1 Chemical compound CCN1CCN(Cc2ccc(COc3ccc([C@H](C(CC[C@@H](c(cc4)ccc4F)O)C4=O)N4c(cc4)ccc4F)c(O)c3)cc2)CC1 COMSZISWKMWFBA-VYOXFEDNSA-N 0.000 description 1
- KEPSBWLVQOJVPV-UHFFFAOYSA-O CCN1CC[NH+](CC(N(CC2)CCN2C(NCc(cc2)ccc2N(C(C2CCC(c(cc3)ccc3F)O)c(ccc(OC)c3)c3O)C2=O)=O)=O)CC1 Chemical compound CCN1CC[NH+](CC(N(CC2)CCN2C(NCc(cc2)ccc2N(C(C2CCC(c(cc3)ccc3F)O)c(ccc(OC)c3)c3O)C2=O)=O)=O)CC1 KEPSBWLVQOJVPV-UHFFFAOYSA-O 0.000 description 1
- NCCSWHYLKAFPTH-BUVRLJJBSA-N COc1cc(OCc2ccccc2)c(/C=N/c2ccc(CN)cc2)cc1 Chemical compound COc1cc(OCc2ccccc2)c(/C=N/c2ccc(CN)cc2)cc1 NCCSWHYLKAFPTH-BUVRLJJBSA-N 0.000 description 1
- KWSPYUOBNIMILB-SANMLTNESA-N C[C@](Cc(cc1)ccc1OCCc1c(C)[o]c(-c2ccc[s]2)n1)(C(O)=O)Oc1ccccc1 Chemical compound C[C@](Cc(cc1)ccc1OCCc1c(C)[o]c(-c2ccc[s]2)n1)(C(O)=O)Oc1ccccc1 KWSPYUOBNIMILB-SANMLTNESA-N 0.000 description 1
- YDBLKRPLXZNVNB-UHFFFAOYSA-N Cc1c(CSc(cc2)cc(C)c2OCC(O)=O)[s]c(-c2ccc(C(F)(F)F)cc2)n1 Chemical compound Cc1c(CSc(cc2)cc(C)c2OCC(O)=O)[s]c(-c2ccc(C(F)(F)F)cc2)n1 YDBLKRPLXZNVNB-UHFFFAOYSA-N 0.000 description 1
- DTDMESKZQRPVRK-UXHLAJHPSA-N Fc(cc1)ccc1/N=C/c(ccc(COCc1ccccc1)c1)c1OCc1ccccc1 Chemical compound Fc(cc1)ccc1/N=C/c(ccc(COCc1ccccc1)c1)c1OCc1ccccc1 DTDMESKZQRPVRK-UXHLAJHPSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/06—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D205/08—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with one oxygen atom directly attached in position 2, e.g. beta-lactams
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
Definitions
- the invention relates to hydroxyl-substituted diphenylazetidinones and their physiologically acceptable salts.
- the object of the invention was to provide further compounds which display a therapeutically utilizable hypolipidemic action.
- the object was to find new compounds which have lower liver levels than the compounds described in the prior art. The lower liver levels reduce the burden on the liver and reduce the possibility of drug-drug interactions.
- the invention therefore relates to compounds of the formula I,
- _C C-, -N ((Ci-C 6) -alkyl) -, -N (phenyl) -, -N ((C 1 -C 6) -alkyl-phenyl) -, -N (CO- (CH 2) i.io-COOH) -, -N (CO- (Ci-C8) -alkyl) -, -N (CO- (C 3 -C 8) cycloalkyl), N (CO- (CH 2) 0 -io-aryl), -N (CO- (CH 2 ) 0- io-heteroaryl), -NH- or by aryl or heteroaryl radicals substituted up to three times by R 7 or by up to four times substituted by R 7 (C 3 - Ci 0 ) -Cycloalkyl or
- Heterocycloalkylreste can be replaced
- a radical or C 2 -C 10 -aliphatic radical having 2 to 10 hydroxyl-substituted radicals, wherein in each case one or more hydroxy functions can be replaced by a -NHR 8 radical; Amino acid residue, oligopeptide residue consisting of 2 to 9 amino acids; acyclic, mono- or bi-cyclic trialkylammonium radical, acyclic, mono- or bicyclic trialkylammoniumalkyl radical, where up to three carbon atoms may be replaced by N, O or S (O), where n 0-2; N-alkylated heteroaromatics, such as. Imidazolium or pyridinium;
- LAG alkylene
- R 7 substituted (C 3 -C 10 ) cycloalkyl or heterocycloalkyl radicals may be replaced.
- group LAG is a sulfate radical (-O-SO 3 H), a sulfonic acid radical (-SO 3 H), a mono- or bicyclic cycloalkyl radical in which one or more carbons are replaced by nitrogen or is a mono- or bicyclic trialkylammoniumalkyl radical.
- a mono- or bicyclic trialkylammonium radical is meant a mono- or bicyclic cycloalkyl radical in which one or more carbons are replaced by nitrogen and the nitrogen carries an additional hydrogen and positive charge.
- a mono- or bicyclic trialkylammoniumalkyl radical is understood as meaning a monocyclic or bicyclic cycloalkyl radical in which one or more carbons are replaced by nitrogen and the nitrogen carries an additional alkyl radical and positive charge. For example, leftovers like
- Alkieinen straight or branched alkyl radical having 1 to 20 carbon atoms Alkieinen straight or branched alkyl radical having 1 to 20 carbon atoms.
- n can be 0-10 and Alki is a straight or branched alkyl radical having 1 to 20 carbon atoms.
- Suitable pharmaceutically acceptable acid addition salts of the compounds according to the invention are salts of inorganic acids, such as hydrochloric acid, hydrobromic acid, phosphoric acid, metaphosphoric acid, nitric acid, sulfonic acid and sulfuric acid, and also organic acids, such as, for example, acetic acid,
- the chloro salt is used in a particularly preferred manner.
- Suitable pharmaceutically acceptable basic salts are ammonium salts, alkali metal salts (such as sodium and potassium salts), alkaline earth salts (such as magnesium and calcium salts), zinc salts, trometamol (2-amino-2-hydroxymethyl-1,3-propanediol), diethanolamine, lysine , Arginine, choline, meglumine or ethylenediamine salts.
- Salts with a non-pharmaceutically acceptable anion or cation are also within the scope of the invention as useful intermediates for the preparation or purification of pharmaceutically acceptable salts and / or for use in non-therapeutic, for example, in vitro applications.
- prodrugs of the compounds of the invention can be metabolized in vivo to a compound of the invention. These prodrugs may or may not be effective.
- the compounds of the invention may also be in various polymorphic forms, e.g. as amorphous and crystalline polymorphic forms. All polymorphic forms of the compounds of the invention are within the scope of the invention and are a further aspect of the invention.
- the compound (s) of the formula (I) can also be administered in combination with other active substances.
- the amount of a compound of formula (I) required to achieve the desired biological effect is dependent upon a number of factors, eg, the specific compound chosen, the intended use, the mode of administration, and the clinical condition of the patient ,
- the daily dose is in the range of 0.1 mg to 100 mg (typically 0.1 mg and 50 mg) per day per kilogram of body weight, eg 0.1-10 mg / kg / day.
- tablets or capsules may contain from 0.01 to 100 mg, typically from 0.02 to 50 mg.
- the abovementioned weights are based on the weight of the diphenylazetidinone ion derived from the salt.
- the compounds according to formula (I) may themselves be used as compound, but they are preferably present with a tolerable carrier in the form of a pharmaceutical composition.
- the carrier must of course be compatible in the sense that it is compatible with the other ingredients of the composition and is not harmful to the patient.
- the carrier may be a solid or a liquid, or both, and is preferably formulated with the compound as a single dose, for example, as a tablet, which may contain from 0.05% to 95% by weight of the active ingredient.
- Other pharmaceutically active substances may also be present, including further compounds according to formula (I).
- the pharmaceutical compositions of the invention can be prepared by one of the known pharmaceutical methods, which consist essentially in that the ingredients with pharmacologically acceptable carriers and / or excipients be mixed.
- compositions according to the invention are those which are suitable for oral and peroral (eg sublingual) administration, although the most suitable mode of administration in each individual case is dependent on the nature and severity of the condition to be treated and on the nature of the particular compound of formula (I) used , Also coated formulations and coated slow release formulations are within the scope of the invention. Preference is given to acid and enteric formulations. Suitable enteric coatings include cellulose acetate phthalate, polyvinyl acetate phthalate,
- Hydroxypropylmethylcellulosephthalat and anionic polymers of methacrylic acid and methyl methacrylate are examples of hydroxypropylmethylcellulosephthalat and anionic polymers of methacrylic acid and methyl methacrylate.
- Suitable pharmaceutical compounds for oral administration may be in separate units, such as capsules, cachets,
- Lozenges or tablets each containing a certain amount of the compound of formula (I); as a powder or granules; as a solution or suspension in an aqueous or non-aqueous liquid; or as an oil-in-water or water-in-oil emulsion.
- these compositions can be prepared by any suitable pharmaceutical method which has a
- the active ingredient and the carrier (which may consist of one or more additional ingredients) are brought into contact.
- the compositions are prepared by uniformly and homogeneously mixing the active ingredient with a liquid and / or finely divided solid carrier, after which the product is molded, if necessary.
- a tablet can be made by compressing or molding a powder or granules of the compound, optionally with one or more additional ingredients.
- Compressed tablets may be prepared by tableting the compound in free-flowing form, such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent and / or surfactant / dispersing agent in a suitable machine.
- Shaped tablets can by molding the powdered, moistened with an inert liquid diluent compound can be prepared in a suitable machine.
- compositions suitable for peroral (sublingual) administration include lozenges containing a compound of formula (I) having a flavor, usually sucrose and gum arabic or tragacanth, and lozenges containing the compound in an inert base such as gelatin and glycerol or sucrose and gum arabic.
- Active ingredient combination can be carried out either by separate administration of the active ingredients to the patient or in the form of combination preparations in which several active ingredients are present in a pharmaceutical preparation.
- Most of the drugs listed below are disclosed in USP Dictionary of USAN and International Drug Names, US Pharmacopeia, Rockville 2001.
- Antidiabetics include insulin and insulin derivatives, such as Lantus ® (see www.lantus.com) or HMR 1964 or those as described in WO2005005477 (Novo Nordisk) fast-acting insulins (see US 6,221, 633), inhalable insulins such as, z. B. Exubera ® or oral insulins such as. B. IN-105 (Nobex) or Oral-lyn TM (Generex Biotechnology), GLP-1 derivatives such as exenatides,
- the orally active hypoglycemic agents preferably comprise sulfonylureas
- Glucokinase activators inhibitors of fructose-1, 6-bisphosphatase
- GLP-1 agonists e.g. those described in WO 97/26265 and
- WO 99/03861 by Novo Nordisk A / S discloses inhibitors of dipeptidyl peptidase-IV (DPP-IV),
- PTP1B protein tyrosine phosphatase 1 B
- Modulators of the sodium-dependent glucose transporter 1 or 2 (SGLT1, SGLT2), fat metabolism-altering compounds such as antihyperlipidemic agents and antilipidemic agents,
- HMGCoA reductase inhibitor such as simvastatin, fluvastatin, Pravastatin, lovastatin, atorvastatin, cerivastatin, rosuvastatin, L-659699.
- the compound of the formula I is administered in combination with a cholesterol absorption inhibitor, such as e.g. Ezetimibe, Tiqueside, Pamaqueside, FM-VP4 (sitostanol / campesterol ascorbyl phosphate; Forbes Medi-Tech, WO2005042692), MD-0727 (Microbia Inc., WO2005021497) or with compounds as in WO2002066464 (Kotobuki Pharmaceutical Co. Ltd.) or WO2005062824 (Merck & Co.) or WO2005061451 and WO2005061452 (AstraZeneca AB).
- a cholesterol absorption inhibitor such as e.g. Ezetimibe, Tiqueside, Pamaqueside, FM-VP4 (sitostanol / campesterol ascorbyl phosphate; Forbes Medi-Tech, WO2005042692), MD-0727 (Microbia Inc., WO2005021497) or with compounds as in WO2002066464 (Ko
- the compound of the formula I is administered in combination with a PPAR gamma agonist, such as e.g. Rosiglitazone, pioglitazone, JTT-501, Gl 262570, R-483, CS-011 (rivoglitazone).
- a PPAR gamma agonist such as e.g. Rosiglitazone, pioglitazone, JTT-501, Gl 262570, R-483, CS-011 (rivoglitazone).
- the compound of formula I is used in combination with PPAR alpha agonist, e.g. GW9578, GW-590735, K-111, LY-674, KRP-101, DRF-10945.
- PPAR alpha agonist e.g. GW9578, GW-590735, K-111, LY-674, KRP-101, DRF-10945.
- the compound of formula I is used in combination with a mixed PPAR alpha / gamma agonist, e.g. Muraglitazar, Tesaglitazar, Naveglitazar, LY-510929, ONO-5129, E-3030, AVE 8042, AVE 8134, AVE 0847, or as in PCT / US 00/11833, PCT / US 00/11490, DE10142734.4 or in JP Berger et al., TRENDS in Pharmacological Sciences 28 (5), 244-251, 2005.
- a mixed PPAR alpha / gamma agonist e.g. Muraglitazar, Tesaglitazar, Naveglitazar, LY-510929, ONO-5129, E-3030, AVE 8042, AVE 8134, AVE 0847, or as in PCT / US 00/11833, PCT / US 00/11490, DE10142734.4 or in JP Berg
- the compound of formula I is administered in combination with a PPAR delta agonist such as GW-501516. In one embodiment, the compound of formula I is administered in combination with metaglidases or with MBX-2044 or other partial PPAR gamma agonist / antagonist In one embodiment of the invention, the compound of formula I is administered in combination with a fibrate, such as fenofibrate, clofibrate, bezafibrate.
- the compound of formula I is administered in combination with an MTP inhibitor, e.g. Implitapide, BMS-201038, R-103757 or those described in WO2005085226.
- an MTP inhibitor e.g. Implitapide, BMS-201038, R-103757 or those described in WO2005085226.
- the compound of formula I is used in combination with a CETP inhibitor, e.g. Torcetrapib or JTT-705.
- a CETP inhibitor e.g. Torcetrapib or JTT-705.
- the compound of formula I is used in combination with bile acid resorption inhibitor (see, for example, US 6,245,744, US 6,221,897 or WO00 / 61568), e.g. HMR 1741 or as described in DE 10 2005 033099.1 and DE 10 2005 033100.9.
- the compound of formula I is used in combination with a polymeric bile acid adsorber, e.g. Cholestyramine, colesevelam.
- a polymeric bile acid adsorber e.g. Cholestyramine, colesevelam.
- the compound of formula I is used in combination with an LDL receptor inducer (see US 6,342,512), e.g. HMR1171, HMR1586, or those as described in WO2005097738.
- an LDL receptor inducer see US 6,342,512
- the compound of formula I is administered in combination with Omacor® (omega-3 fatty acids, high-concentration ethyl esters of eicosapentaenoic acid and docosahexaenoic acid).
- Omacor® omega-3 fatty acids, high-concentration ethyl esters of eicosapentaenoic acid and docosahexaenoic acid.
- the compound of formula I is administered in combination with an ACAT inhibitor, e.g. Avasimibe, administered.
- an ACAT inhibitor e.g. Avasimibe
- the compound of the formula I is described in Combination with an antioxidant, such as OPC-14117, probucol, tocopherol, ascorbic acid, ß-carotenes or selenium.
- an antioxidant such as OPC-14117, probucol, tocopherol, ascorbic acid, ß-carotenes or selenium.
- the compound of the formula I in combination with a vitamin, such as. As vitamin B6 or vitamin B12 administered.
- the compound of the formula I is administered in combination with a lipoprotein-lipase modulator, e.g. Ibrolipim (NO-1886).
- a lipoprotein-lipase modulator e.g. Ibrolipim (NO-1886).
- the compound of formula I is used in combination with an ATP citrate lyase inhibitor, e.g. SB-204990 administered.
- an ATP citrate lyase inhibitor e.g. SB-204990 administered.
- the compound of formula I is used in combination with a squalene synthetase inhibitor, e.g. BMS-188494 or as described in WO2005077907.
- a squalene synthetase inhibitor e.g. BMS-188494 or as described in WO2005077907.
- the compound of formula I in combination with a lipoprotein (a) antagonist such as e.g. Gemcabene (CI-1027).
- the compound of formula I in combination with an HM74A receptor agonist such as e.g. Nicotinic acid administered.
- the compound of formula I is administered in combination with a lipase inhibitor, e.g. Orlistat or cetilistat (ATL-962).
- a lipase inhibitor e.g. Orlistat or cetilistat (ATL-962).
- the compound of the formula I is administered in combination with insulin. In one embodiment, the compound of formula I is administered in combination with a sulfonylurea such as tolbutamide, glibenclamide, glipizide or glimepiride.
- a sulfonylurea such as tolbutamide, glibenclamide, glipizide or glimepiride.
- the compound of formula I is used in combination with a biguanide, e.g. Metformin, administered.
- a biguanide e.g. Metformin
- the compound of formula I is used in combination with a meglitinide, e.g. Repaglinide or nateglinide.
- a meglitinide e.g. Repaglinide or nateglinide.
- the compound of formula I is used in combination with a thiazolidinedione, e.g. Troglitazone, ciglitazone, pioglitazone, rosiglitazone or those described in WO 97/41097 by Dr. med. Reddy's Research Foundation disclosed compounds, particularly 5 - [[4 - [(3,4-dihydro-3-methyl-4-oxo-2-quinazolinylmethoxy) phenyl] methyl] -2,4-thiazolidinedione.
- the compound of formula I is administered in combination with an ⁇ -glucosidase inhibitor, e.g. Miglitol or acarbose, administered.
- an ⁇ -glucosidase inhibitor e.g. Miglitol or acarbose
- the compound of formula I is administered in combination with an agent which acts on the ATP-dependent potassium channel of beta cells, e.g. Tolbutamide, glibenclamide, glipizide, glimepiride or repaglinide.
- an agent which acts on the ATP-dependent potassium channel of beta cells e.g. Tolbutamide, glibenclamide, glipizide, glimepiride or repaglinide.
- the compound of formula I is used in combination with more than one of the aforementioned compounds, e.g. in combination with a sulphonylurea and metformin, a sulphonylurea and acarbose, repaglinide and metformin, insulin and a sulphonylurea, insulin and metformin, insulin and troglitazone, insulin and lovastatin, etc.
- the compound of the formula I is administered in combination with an inhibitor of glycogen phosphorylase, such as, for example, PSN-357 or FR-258900 or those as described in WO2003084922, WO2004007455, WO2005073229-31, WO2005067932.
- the compound of formula I is administered in combination with glucagon receptor antagonists such as A-770077 or NNC-25-2504 or as described in WO2004100875, WO2005065680.
- the compound of the formula I in combination with activators of glucokinase such as. LY-2121260 (WO2004063179), PSN-105, PSN-110, GKA-50, or those as described e.g. As described in WO2004072031 or WO2004072066 or WO2005080360.
- the compound of the formula I in combination with an inhibitor of gluconeogenesis, such as. FR-225654.
- the compound of formula I is used in combination with inhibitors of fructose-1, 6-bisphosphatase (FBPase), e.g. CS-917 administered.
- FBPase 6-bisphosphatase
- the compound of formula I in combination with modulators of glucose transporter-4 (GLUT4), such as. KST-48 (D.O. Lee et al .: Arzneim.-Forsch. Drug Res. 54 (12), 835 (2004)).
- the compound of the formula I is used in combination with
- Inhibitors of glutamine-fructose-6-phosphate amidotransferase as described e.g. As described in WO2004101528 administered.
- the compound of formula I in combination with inhibitors of dipeptidyl peptidase-IV such as. Vildagliptin (LAF-237),
- Sitagliptin MK-0431
- saxagliptin (BMS-477118)
- GSK-823093 PSN-9301
- SYR-322 SYR-619
- TA-6666 TS-021
- GRC-8200 GW-825964X or as described in U.S.
- the compound of formula I in combination with inhibitors of protein tyrosine phosphatase-1 B (PTP1 B), as z.
- PTP1 B protein tyrosine phosphatase-1 B
- the compound of formula I is used in combination with modulators of the sodium-dependent glucose transporter 1 or 2 (SGLT1, SGLT2), e.g. KGA-2727, T-1095, SGL-0010, AVE 2268 and SAR 7226 or as described e.g. In WO2004007517, WO200452903, WO200452902, PCT / EP2005 / 005959,
- the compound of formula I is administered in combination with modulators of GPR40.
- the compound of the formula I in combination with inhibitors of hormone-sensitive lipase (HSL), such.
- HSL hormone-sensitive lipase
- the compound of the formula I in combination with inhibitors of acetyl-CoA carboxylase (ACC) such. B. such as in W0199946262, WO200372197, WO2003072197, WO2005044814.
- the compound of formula I is used in combination with an inhibitor of phosphoenolpyruvate carboxykinase (PEPCK), e.g. such as described in WO2004074288 administered.
- PPCK phosphoenolpyruvate carboxykinase
- the compound of the formula I in combination with an inhibitor of glycogen synthase kinase-3 beta in combination with an inhibitor of glycogen synthase kinase-3 beta (GSK-3 beta), such as.
- an inhibitor of glycogen synthase kinase-3 beta such as.
- GSK-3 beta glycogen synthase kinase-3 beta
- GSK-3 beta glycogen synthase kinase-3 beta
- the compound of formula I in combination with an inhibitor of protein kinase C beta such as. B. Ruboxistaurin administered.
- PLC beta protein kinase C beta
- the compound of formula I in combination with an endothelin A receptor antagonist, such as. B. avosentan (SPP-301).
- an endothelin A receptor antagonist such as. B. avosentan (SPP-301).
- the compound of the formula I is administered in combination with inhibitors of the "1-kappaB kinase" (IKK inhibitors), as described, for example, in WO2001000610, WO2001030774, WO2004022553, WO2005097129.
- IKK inhibitors inhibitors of the "1-kappaB kinase”
- the compound of the formula I in combination with modulators of the glucocorticoid receptor, as described, for. As described in WO2005090336 administered.
- the compound of the formula I is used in combination with CART modulators (see “cocaine-amphetamine-regulated transcript-influenced transient-energy metabolism, anxiety and gastric emptying in mice" Asakawa, A. et al .: Hormone and Metabolism Research (2001 ), 33 (9), 554-558); NPY antagonists such as naphthalene-1-sulfonic acid ⁇ 4 - [(4-amino-quinazolin-2-ylamino) -methyl] -cyclohexylmethyl ⁇ -amide hydrochloride (CGP 71683A); Peptide YY 3-36 (PYY3-36) or analogous compounds such.
- CJC-1682 PYY3-36 conjugated to human serum albumin via Cys34
- CJC-1643 derivative of PYY3-36 conjugated to serum albumin in vivo
- Cannabinoid receptor 1 antagonists such as rimonabant, SR147778 or those as described, for example, in EP 0656354, WO 00/15609, WO 02/076949, WO2005080345, WO2005080328, WO2005080343, WO2005075450, WO2005080357, WO200170700, WO2003026647-48, WO200302776, WO2003040107,
- WO2005063761-62, WO2005061509, WO2005077897 are described); MC4 agonists (eg 1-amino-1,2,3,4-tetrahydronaphthalene-2-carboxylic acid [2- (3-benzyl-2-methyl-3-oxo-2,3,3a, 4,6, 7-hexahydro-pyrazolo [4,3-c] pyridin-5-yl) -1- (4-chlorophenyl) -2-oxo-ethyl] -amide (WO 01/91752) or LB53280, LB53279, LB53278 or THIQ, MB243, RY764, CHIR-785, PT-141 or those as described in WO2005060985, WO2005009950, WO2004087159, WO2004078717, WO2004078716, WO2004024720, US20050124652, WO2005051391, WO2004112793, WO20020022
- WO200501921, WO200509184, WO2005000339, EP1460069, WO2005047253, WO2005047251, EP1538159, WO2004072076, WO2004072077 are described; Orexin receptor antagonists (eg, 1- (2-methyl-benzoxazol-6-yl) -3- [1,5] naphthyridin-4-yl-urea hydrochloride (SB-334867-A) or those as described, for example, in US Pat in WO200196302, WO200185693, WO2004085403, WO2005075458); Histamine H3 receptor agonists (eg, 3-cyclohexyl-1- (4,4-dimethyl-1,4,6,7-tetrahydro-imidazo [4,5-c] pyridin-5-yl) -propane-1 oxalic acid salt (WO 00/63208) or those as described in WO200064884, WO2005082893); CRF antagonist
- 5-HT receptor agonists e.g. 1- (3-ethyl-benzofuran-7-yl) -piperazine oxalic acid salt (WO 01/09111);
- 5-HT2C receptor agonists such as APD-356 or BVT-933 or those as described in WO200077010, WO20077001-02, WO2005019180, WO2003064423, WO200242304, WO2005082859);
- 5-HT6 receptor antagonists as described, for example, in WO2005058858; Bombesin receptor agonists (BRS-3 agonists; Galanin receptor antagonists;
- Growth hormone e.g., human growth hormone or AOD-9604
- human growth hormone e.g., human growth hormone or AOD-9604
- Ghrelin antagonists such as
- TRH agonists see, e.g., EP 0 462 884; decoupling protein 2- or 3-modulators; Leptin agonists (see, e.g., Lee, Daniel W., Leinung, Matthew C; Rozhavskaya;
- DA agonists bromocriptine, doprexin
- Lipase / amylase inhibitors e.g., WO 00/40569
- Inhibitors of diacylglycerol O-acyltransferases DGATs
- FAS fatty acid synthase
- thyroid hormone receptor agonists such as. B: KB-2115 or those as described in WO20058279, WO200172692, WO200194293, WO2003084915, WO2004018421, WO2005092316.
- the further active ingredient is leptin; see eg "Perspectives in the Therapeutic Use of Leptin", Salvador, Javier; Gomez-Ambrosi, Javier; Fruhbeck, Gema, Expert Opinion on Pharmacotherapy (2001), 2 (10), 1615-1622.
- the other active ingredient is dexamphetamine or amphetamine.
- the other active ingredient is fenfluramine or dexfenfluramine.
- the other active ingredient is sibutramine.
- the other active ingredient is mazindol or phentermine.
- the compound of formula I in combination with bulking agents preferably insoluble bulking agents
- bulking agents preferably insoluble bulking agents
- Caromax is a carob-containing product of the company Nutrinova, Nutrition Specialties & Food Ingredients GmbH, Industriepark availability, 65926 Frankfurt / Main)) administered.
- Combination with Caromax ® is possible in one preparation or by separate administration of compounds of the formula I and Caromax ®.
- Caromax ® can also be administered in the form of food, such as in baked goods or muesli bars.
- the invention furthermore relates to stereoisomer mixtures of the formula I as well as to the pure stereoisomers of the formula I and to diastereomer mixtures of the formula I and also to the pure diastereomers.
- the separation of the mixtures is carried out by chromatographic means.
- Preferred amino-protecting groups are the benzyloxycarbonyl (Z) radical which can be split off by catalytic hydrogenation, the 2- (3,5-dimethyloxyphenyl) propyl (2) oxycarbonyl (Ddz) or trityl (Trt) radical which can be split off by weak acids t-butylcarbamate (BOC) radical which can be split off by acids, such as 3M hydrochloric acid, and the 9-fluorenylmethyloxycarbonyl (Fmoc) radical cleavable by secondary amines.
- Z benzyloxycarbonyl
- Ddz 2- (3,5-dimethyloxyphenyl) propyl (2) oxycarbonyl
- Trt trityl
- BOC t-butylcarbamate
- Fmoc 9-fluorenylmethyloxycarbonyl
- the invention further relates to a process for the preparation of diphenylazetidinone derivatives of the formula I.
- linkage of - (CH 2 ) oiYWZ- (Co-C 25 ) -alkylene-H in compound II can alternatively also be on one of the other two phenyl rings.
- the organic phase is filtered through 100 ml of silica gel.
- the aqueous phase is extracted again with 80 ml of n-heptane / ethyl acetate (2: 1) and the organic phase is used to wash the silica gel of the first filtration.
- the organic phase is concentrated and purified by flash chromatography (n-heptane / ethyl acetate 4: 1 to 2: 1). 4.34 g of product 12 are obtained.
- Example 19 38 mg (0.075 mmol) of compound 19 are reacted analogously to the synthesis of Example A and 30 mg of Example B are obtained as an amorphous solid having a molecular weight of 587.11 (C 29 H 3I F 2 N 3 O 6 S); MS (ESI ' ): 586.11 (MH " ).
- Piperazine-1-carbonic acid (4- [3- [3- (tert-butyl-dimethyl-silanyloxy) -3- (4-fluorophenyl-1-propyl-1- (4-fluorophenvl) -4-oxo] azetidine-2-vll-3-hydroxy-2-dibenzyl) -amide 28
- aniline 27 700 mg (1.3 mmol) of aniline 27 are reacted analogously to the synthesis of amine 15 and 347 mg of 28 are obtained as an amorphous solid.
- Example E 370 mg (0.69 mmol) of compound 29 are reacted analogously to the synthesis of Example A and 343 mg of sulfuric acid amide 30 (Example E) are obtained as amorphous solid having the molecular weight 616.18 (C 29 H 30 F 2 N 4 O 7 S); MS (ESI + ): 1233.28 (2M + H + ).
- Example H 54 mg (0.13 mmol) of aniline 31 and 150 mg of iodoacetic acid are coupled in a similar manner to EDC / HOBt as described in the synthesis of Ex. G and 40 mg of iodide are obtained. This is dissolved in 5 ml of toluene and 1 ml of methylene chloride. After addition of 200 ml of DABCO is stirred at 80 0 C for 1 hour. The precipitate is filtered off with suction and the trialkylammonium alkyl salt Ex. H is obtained with the molecular weight 577.26 (C 32 H 35 F 2 N 4 O 4 ); MS (ESI): 577.20 (M + ).
- phenol 37 0.5 g (0.79 mmol) of phenol 37 are alkylated with 700 mg (2.4 mmol) of iodide 46 and 650 mg of K 2 CO 3 in 15 ml of DMF analogously to the synthesis of compound 44, giving 300 mg of 47 as an amorphous solid.
- 150 mg (0.25 mmol) of 48 are dissolved in 2 ml of pyridine. After addition of 170 mg trimethylamine sulfur trioxide complex is stirred for 1 hour at room temperature. Then it is diluted with 10 ml of methanol and 10 ml of toluene and concentrated. The residue is columned by flash chromatography (methylene chloride / methanol / concentrated ammonia 30/5/1 then 30/10/3) to give 150 mg of sulphate 49.
- Example L having the molecular weight 577.19 (C 28 H 29 F 2 NO 8 S); MS (ESI + ): 578.26 (M + H + ).
- the animals are labeled 24 hours before the oral administration of the test meal ( 14 C-cholesterol in Intralipid® 20, Pharmacia-Upjohn) with 3 H-TCA (taurocholic acid) sc (eg 1 ⁇ Ci / mouse to 5 ⁇ Ci / rat).
- 3 H-TCA taurocholic acid
- Cholesterol absorption test 0.25 ml / mouse Intralipid® 20 (Pharmacia-Upjohn) ((spiked with 0.25 ⁇ Ci 14 C-cholesterol in 0.1 mg cholesterol) are administered orally by gavage. Test substances are prepared separately in 0.5% / (methylcellulose (Sigma) / 5% Solutol (BASF, Ludwigshafen) or suitable vehicle.) The application volume of the test substance is 0.5 ml / mouse The test substance is administered immediately before the test meal (intralipid with 14 C-cholesterol label) (cholesterol absorption test) administered orally.
- the feces are collected over 24 h: the fecal elimination of 14 C-cholesterol and 3 H taurocholic acid (TCA) after 24 h is determined.
- the livers are harvested, homogenized and aliquots in oxime (Model 307, P Paacckkaarrdd) are used to determine the ingested / absorbed amount of 14 C-cholesterol.
- the amount of 14 C-cholesterol absorbed into the liver is evaluated as a function of the administered dose of the test substance in relation to a control group (vehicle-treated).
- the ED 50 values are interpolated from a dose response curve as the dose that halves (50%) the uptake of 14 C-cholesterol into the liver relative to a control group.
- the following ED 50 values (liver values, mouse) substantiate the activity of the compounds of the formula I according to the invention
- test substances are prepared in 0.5% / (methylcellulose (Sigma) / 5% Solutol (BASF, Ludwigshafen) or another suitable vehicle and administered at least 3 doses on 12 consecutive days once a day with the gavage Animals in deep anesthesia from the aorta are bled in.
- the serum contains total cholesterol, LDL cholesterol, HDL cholesterol and triglycerides with standard Roche kits according to the guidelines of the German Society for Clinical Chemistry analyzed.
- the ED 50 values for LDL cholesterol cholesterol lowering versus placebo-treated control animals were calculated using a standard logistic model for the dose-response curve.
- ED 50 values serum LDL cholesterol values, hamster, [mg / kg] demonstrate the activity of the compounds of the formula I according to the invention:
- the liver exposure of the compounds according to the invention to the corresponding compounds without hydroxy function in the 2 "position was investigated in vivo on the male Wistar rat, on the rat anesthetized with ketamine / midazolam (ketamine 80 mg / kg ip + midazolam 5 mg / kg ip)
- the preparation is administered intraduodenally, attempting to prevent immediate reflux of the preparation into the stomach.
- the animals remain anesthetized throughout the experiment, and at the end of the experiment, after 2 h, the liver becomes the substance
- the determination of the substance levels in the liver homogenates is carried out by LC-MS / MS by first precipitating the proteins by adding acetonitrile in the presence of an internal standard, adding a suitable buffer to a portion of the supernatant and making an aliquot of the mixture The evaluation of the measurement takes place via the peak areas of analyte and i standard.
Abstract
Die Erfindung betrifft Verbindungen der Formel (I), worin R1, R2, R3, R4, R5, und R6 die angegebenen Bedeutungen haben, sowie deren physiologisch verträgliche Salze. Die Verbindungen eignen sich z.B. als Hypolipidämika.
Description
Beschreibung
HYDROXY-SUBSTITUIERTE DIPHENYLAZETTDINONE ZUR BEHANDLUNG VON HYPERLIPIDÄMIE
Die Erfindung betrifft mit Hydroxyfunktionen substituierte Diphenylazetidinone und deren physiologisch verträgliche Salze.
Es sind bereits gering resorbierebare Diphenylazetidinone sowie deren Verwendung zur Behandlung von Hyperlipidämie beschrieben worden (PCT/EP03/05816, PCT/EP03/05815 und PCT/EP03/05816).
Der Erfindung lag die Aufgabe zugrunde, weitere Verbindungen zur Verfügung zu stellen, die eine therapeutisch verwertbare hypolipidämische Wirkung entfalten. Insbesondere bestand die Aufgabe darin, neue Verbindungen zu finden, die gegenüber den im Stand der Technik beschriebenen Verbindungen, geringere Leberspiegel aufweisen. Die geringeren Leberspiegel reduzieren die Belastung der Leber und vermindern die Möglichkeit von Drug-Drug Interaktionen.
Die Aufgabe wird gelöst durch Verbindungen, die eine zusätzliche Hydroxyfunktion in 2"-Stellung tragen.
Die Erfindung betrifft daher Verbindungen der Formel I,

R1 , R2, R3, R4, R5, R6 unabhängig voneinander (Ci-C3o)-Alkylen-(LAG)n, wobei n = 1 - 5 sein kann und wobei ein oder mehrere C-Atome des Alkylenrests durch -S(O)n-, mit n = 0 - 2, -O-, -(C=O)-, -(C=S)-, -CH=CH-,
_C=C-, -N((Ci-C6)-Alkyl)-, -N(Phenyl)-, -N((C1-C6)-Alkyl-Phenyl)-, -N(CO- (CH2)i.io-COOH)-, -N(CO-(Ci -C8)-Alkyl)-, -N(CO-(C3-C8)-Cycloalkyl), N(CO-(CH2)0-io-Aryl), -N(CO-(CH2)0-io-Heteroaryl), -NH- oder durch bis zu dreifach mit R7 substituierte Aryl- oder Heteroarylreste oder durch bis zu vierfach mit R7 substituierte (C3-Ci0)-Cycloalkyl- oder
Heterocycloalkylreste ersetzt sein können;
H, F, Cl, Br, I, CF3, NO2, N3, CN, COOH, COO(CrC6)Alkyl, CONH2, CONH(Ci-C6)Alkyl, CON[(Ci-C6)Alkyl]2, (CrC6)-Alkyl, (C2-C6)-Alkenyl, (C2- C6)-Alkinyl, O-(Ci-C6)-Alkyl, wobei in den Alkylresten ein, mehrere, oder alle Wasserstoff(e) durch Fluor ersetzt sein können; C(=NH)(NH2), PO3H2, SO3H, SO2-NH2, SOzNH^-CeJ-Alkyl, SO2Nf(C1- C6)-Alkyl]2 , S-(C1-C6)-Alkyl, S-(CH2)n-Phenyl, SO-(C1-C6)-Alkyl, SO- (CH2)n-Phenyl, SO2-(Ci-C6)-Alkyl, SO2-(CH2)n-Phenyl, wobei n = O - 6 sein kann und der Phenylrest bis zu zweifach mit F, Cl, Br, OH, CF3, NO2,
CN, OCF3, O-(Ci-C6)-Alkyl, (d-C6)-Alkyl, NH2 substituiert sein kann; NH2, NH^d-CeJ-Alkyl, N((Ci-C6)-Alkyl)2, NH(C1-Cy)-ACyI, Phenyl, O- (CH2)n-Phenyl, wobei n = O - 6 sein kann, wobei der Phenylring ein bis 3- fach substituiert sein kann mit F, Cl, Br, I, OH, CF3, NO2, CN, OCF3, O- (d-CβJ-Alkyl, (C1-Ce)-AIk^, NH2, NH(CrC6)-Alkyl, N((C1-C6)-Alkyl)2, SO2-
CH3, COOH, COO-(CrC6)-Alkyl, CONH2;
R7 F, Cl, Br, I, CF3, NO2, N3, CN, COOH, COOtd-QOAlkyl, CONH2,
CONHtd-CeJAlkyl, CON[(C1-C6)Alkyl]2, (CrC6)-Alkyl, (C2-C6)-Alkenyl, (C2- C6)-Alkinyl,
 wobei in den Alkylresten ein, mehrere, oder alle Wasserstoff(e) durch Fluor ersetzt sein können; C(=NH)(NH2), PO3H2, SO3H, SO2-NH2, SOzNHfd-CeJ-Alkyl, SO2Nf(C1-
C6)-Alkyl]2 , S-(Ci-C6)-Alkyl, S-(CH2)n-Phenyl, SO-(CrC6)-Alkyl, SO- (CH2)n-Phenyl, SOHd-Ceϊ-Alkyl, SO2-(CH2)n-Phenyl, wobei n = 0 - 6 sein kann und der Phenylrest bis zu zweifach mit F, Cl, Br, OH, CF3, NO2, CN, OCF3, O-(Ci-C6)-Alkyl, (CrC6)-Alkyl, NH2 substituiert sein kann; NH2, NH-(CrC6)-Alkyl, NKd-CeJ-Alkyl);,, NH(C1-Cr)-ACyI, Aryl, O-(CH2)n-
wobei in den Alkylresten ein, mehrere, oder alle Wasserstoff(e) durch Fluor ersetzt sein können; C(=NH)(NH2), PO3H2, SO3H, SO2-NH2, SOzNHfd-CeJ-Alkyl, SO2Nf(C1-
C6)-Alkyl]2 , S-(Ci-C6)-Alkyl, S-(CH2)n-Phenyl, SO-(CrC6)-Alkyl, SO- (CH2)n-Phenyl, SOHd-Ceϊ-Alkyl, SO2-(CH2)n-Phenyl, wobei n = 0 - 6 sein kann und der Phenylrest bis zu zweifach mit F, Cl, Br, OH, CF3, NO2, CN, OCF3, O-(Ci-C6)-Alkyl, (CrC6)-Alkyl, NH2 substituiert sein kann; NH2, NH-(CrC6)-Alkyl, NKd-CeJ-Alkyl);,, NH(C1-Cr)-ACyI, Aryl, O-(CH2)n-
Aryl, wobei n = O - 6 sein kann, wobei der Arylring ein bis 3-fach substituiert sein kann mit F, Cl, Br, I, OH, CF3, NO2, CN, OCF3, 0-(C1-C6)- Alkyl, (CrC6)-Alkyl, NH2, NH(C1-C6)-Alkyl, N((C1-C6)-Alkyl)2, SO2-CH3, COOH, COO-tCrCeJ-Alkyl, CONH2;
LAG C4-C10-cycloaliphatischer mit 2 bis 9 Hydroxyfunktionen substituierter
Rest oder C2-C10-aliphatischer mit 2 bis 10 Hydroxyfunktionen substituierter Rest, wobei jeweils eine oder mehrere Hydroxyfunktionen durch einen -NHR8-Rest ersetzt sein können; Aminosäurerest, Oligopeptidrest bestehend aus 2 bis 9 Aminosäuren; acyclischer, mono- oder bi-cyclischer Trialkylammonium-Rest, acyclischer, mono- oder bicyclischer Trialkylammoniumalkyl-Rest, wobei bis zu drei Kohlenstoffatome durch N, O oder S(O), mit n = 0-2, ersetzt sein können; N-alkylierte Heteroaromaten, wie z. B. Imidazolium oder Pyridinium;
-O-(SO2)-OH; -(CH2)O-10-SO3H; -(CH2)0-10-P(O)(OH)2, -(CH2)0-10-O- P(O)(OH)2, -(CH2)0-10-C(=NH)(NH2); -(CH2)0-10-C(=NH)(NHOH); -NR8- C(=NR9)(NR10R11); wobei n = 1 - 5 ist und R8, R9, R10 und R11 unabhängig voneinander H, (C-i-CβJ-Alkyl, Phenyl, (CrCe)-Alkyl-Phenyl, (C3-C8)-Cycloalkyl), -C(O)-(C1-C6)-Alkyl, -C(O)-(C3-C8)-Cycloalkyl sein können;
wobei immer mindestens einer der Reste R1 bis R6 die Bedeutung (C1-C30)-Alkylen-(LAG)n, wobei n = 1 - 5 sein kann und wobei ein oder mehrere C- Atome des Alkylenrests durch -S(O)n-, mit n = O - 2, -O-, -(C=O)-, -(C=S)-, -CH=CH-, - C≡C-, -N((C1-C6)-Alkyl)-, -N(Phenyl)-, -N((C1-C6)-Alkyl-Phenyl)-, -N(CO-(CH2)1-10- COOH)-, -N(CO-(CrC8)-Alkyl)-, -N(CO-(C3-C8)-Cycloalkyl), N(CO-(CH2)0-10-Aryl), -
N(CO-(CH2)o-io-Heteroaryl), -NH- oder durch bis zu dreifach mit R7 substituierte Aryl- oder Heteroarylreste oder durch bis zu vierfach mit R7 substituierte (C3-Ci0)- Cycloalkyl- oder Heterocycloalkylreste ersetzt sein können; besitzen muß,
sowie deren pharmazeutisch verträglichen Salze.
Bevorzugt sind Verbindungen der Formel I, worin mindestens einer der Reste R1 bis R6 die Bedeutung (Ci-C2o)-Alkylen-(LAG), wobei ein oder mehrere C-Atome des Alkylenrests durch -O-, -(C=O)-, -N((Ci-C6)-Alkyl)-, -N(CO-(CH2)I-Io-COOH)- oder - NH- oder durch bis zu dreifach mit R7 substituierte Aryl- oder Heteroarylreste oder durch bis zu vierfach mit R7 substituierte (C3-Ci0)-Cycloalkyl- oder Heterocycloalkylreste ersetzt sein können, besitzt.
Besonders bevorzugt sind Verbindungen der Formel I1 worin einer der Reste R1 oder R3 die Bedeutung (Ci-C12)-Alkylen-(LAG) hat, wobei ein oder mehrere C-Atome des Alkylenrests durch -O-, -(C=O)-, -N(CH3)-, oder -NH- oder durch bis zu dreifach mit R7 substituierte Aryl- oder Heteroarylreste oder durch bis zu vierfach mit R7 substituierte (C3-C10)-Cycloalkyl- oder Heterocycloalkylreste ersetzt sein können.
Ganz besonders bevorzugt sind Verbindungen der Formel I, worin einer der Reste R1 oder R3 die Bedeutung (Ci-C5)-Alkylen-(LAG) hat; worin ein oder mehrere C-Atome des Alkylenrests durch -O-, -(C=O)- oder -NH- oder durch bis zu dreifach mit R7 substituierte Aryl- oder Heteroarylreste oder durch bis zu vierfach mit R7 substituierte (C3-C10)-Cycloalkyl- oder Heterocycloalkylreste ersetzt sein können.
Noch weiter bevorzugt sind Verbindungen der Formel I, worin einer der Reste R1 oder R3 die Bedeutung -O-CH2-Aryl-CH2-(LAG), -CH2-O-(C=O)-Heterocycloalkyl-(LAG), - CH2-NH-(C=O)-Heterocycloalkyl-(C=O)-CH2-(LAG), -CH2-Heterocycloalkyl-(LAG), -NH- (C=O)-Heterocycloalkyl-(LAG) oder -O-(C=O)-Heterocycloalkyl-(LAG) besitzt.
Weiter bevorzugt sind Verbindungen der Formel I, worin einer der Reste R1 oder R3
die Bedeutung -CH2-O-(C=O)-Heterocycloaikyl-(LAG), -CH2-NH-(C=O)- Heterocycloalkyl-(C=O)-CH2-(LAG), -CH2-Heterocycloalkyl-(LAG), -NH-(C=O)- Heterocycloalkyl-(LAG) oder -O-(C=O)-Heterocycloalkyl-(LAG) besitzt.
Weiter bevorzugt sind Verbindungen der Formel I, worin einer der Reste R1 oder R3
— N N — als Heterocycloalkylrest den 1 ,4-Piperazindiylrest ( ^^ ) besitzt.
Weiterhin bevorzugt sind Verbindungen der Formel I, worin die Gruppe LAG ein Sulfatrest (-0-SO3H), ein Sulfonsäurerest (-SO3H), ein mono- oder bicyclischer Cycloalkylrest, in dem ein oder mehrere Kohlenstoffe durch Stickstoff ausgetauscht sind oder ein mono- oder bicyclischer Trialkylammoniumalkyl-Rest ist.
Unter einem mono- oder bicyclischen Trialkylammonium-Rest wird ein mono- oder bicyclischer Cycloalkylrest, in dem ein oder mehrere Kohlenstoffe durch Stickstoff ausgetauscht sind und der Stickstoff einen zusätzlichen Wasserstoff und positive Ladung trägt, verstanden.
Z.B. Reste wie
, wobei n, m und p unabhängig voneinander 0 - 10 sein können und eine oder mehrere CH2-Gruppen unabhängig voneinander durch O, S(O)n, wobei n = 0 - 2 sein kann, NH, N-(Ci-CiO)-Alkyl, N-Phenyl oder N-CH2-Phenyl ersetzt sein können.
Unter einem mono- oder bicyclischen Trialkylammoniumalkyl-Rest wird ein mono- oder bicyclischer Cycloalkylrest, in dem ein oder mehrere Kohlenstoffe durch Stickstoff ausgetauscht sind und der Stickstoff einen zusätzlichen Alkylrest und positive Ladung trägt, verstanden.
Z.B. Reste wie

oder

oder

verstanden, wobei n, m und p unabhängig voneinander 0 - 10 sein können und eine oder mehrere CH2-Gruppen unabhängig voneinander durch O, S(O)n, wobei n = 0 - 2 sein kann, NH, N-(C1-CiO)-AIkYl, N-Phenyl oder N-CH2-Phenyl ersetzt sein können und
Alkieinen geraden oder verzweigten Alkylrest mit 1 bis 20 Kohlenstoffatomen bedeutet.
Unter N-alkylierten Heteroaromaten werden Reste wie z.B.
AIk '(CH2)n~

N .
AIk1- 'N ^N' -(CH,)n> AIk;
N^N+ -(CH,)n. Alk; /1V + -(CH2)n -N N
N N: V-=/


verstanden, wobei n 0 - 10 sein kann und Alki einen geraden oder verzweigten Alkylrest mit 1 bis 20 Kohlenstoffatomen bedeutet.
Pharmazeutisch verträgliche Salze sind aufgrund ihrer höheren Wasserlöslichkeit gegenüber den Ausgangs- bzw. Basisverbindungen besonders geeignet für
medizinische Anwendungen. Diese Salze müssen ein pharmazeutisch verträgliches Anion oder Kation aufweisen. Geeignete pharmazeutisch verträgliche Säureadditionssalze der erfindungsgemäßen Verbindungen sind Salze anorganischer Säuren, wie Salzsäure, Bromwasserstoff-, Phosphor-, Metaphosphor-, Salpeter-, Sulfon- und Schwefelsäure sowie organischer Säuren, wie z.B. Essigsäure,
Benzolsulfon-, Benzoe-, Zitronen-, Ethansulfon-, Fumar-, Glucon-, Glykol-, Isethion-, Milch-, Lactobion-, Malein-, Apfel-, Methansulfon-, Bernstein-, p-Toluolsulfon-, und Weinsäure. Für medizinische Zwecke wird in besonders bevorzugter Weise das Chlorsalz verwendet. Geeignete pharmazeutisch verträgliche basische Salze sind Ammoniumsalze, Alkalimetallsalze (wie Natrium- und Kaliumsalze), Erdalkalisalze (wie Magnesium- und Calciumsalze), Zinksalze, Trometamol (2-Amino-2-hydroxymethyl-1 ,3-propandiol)-, Diethanolamin-, Lysin-, Arginin-, Cholin-, Meglumin- oder Ethylendiamin-Salze.
Salze mit einem nicht pharmazeutisch verträglichen Anion oder Kation gehören ebenfalls in den Rahmen der Erfindung als nützliche Zwischenprodukte für die Herstellung oder Reinigung pharmazeutisch verträglicher Salze und/oder für die Verwendung in nicht-therapeutischen, zum Beispiel in-vitro-Anwendungen.
Ein weiterer Aspekt dieser Erfindung sind Prodrugs der erfindungsgemäßen Verbindungen. Solche Prodrugs können in vivo zu einer erfindungsgemäßen Verbindung metabolisiert werden. Diese Prodrugs können selbst wirksam sein oder nicht.
Die erfindungsgemäßen Verbindungen können auch in verschiedenen polymorphen Formen vorliegen, z.B. als amorphe und kristalline polymorphe Formen. Alle polymorphen Formen der erfindungsgemäßen Verbindungen gehören in den Rahmen der Erfindung und sind ein weiterer Aspekt der Erfindung.
Nachfolgend beziehen sich alle Verweise auf "Verbindung(en) gemäß Formel (I)" auf Verbindung(en) der Formel (I) wie vorstehend beschrieben, sowie ihre Salze, Solvate und physiologisch funktionellen Derivate wie hierin beschrieben.
Die Verbindungen der Formel I und deren pharmazeutisch verträgliche Salze und physiologisch funktionelle Derivate stellen ideale Arzneimittel zur Behandlung von Lipidstoffwechselstörungen, insbesondere von Hyperlipidämie dar. Die Verbindungen der Formel I eignen sich ebenfalls zur Beeinflussung des Serumcholesterinspiegels sowie zur Prävention und Behandlung arteriosklerotischer Erscheinungen.
Die Verbindung(en) der Formel (I) können auch in Kombination mit weiteren Wirkstoffen verabreicht werden.
Die Menge einer Verbindung gemäß Formel (I), die erforderlich ist, um den gewünschten biologischen Effekt zu erreichen, ist abhängig von einer Reihe von Faktoren, z.B. der gewählten spezifischen Verbindung, der beabsichtigten Verwendung, der Art der Verabreichung und dem klinischen Zustand des Patienten. Im allgemeinen liegt die Tagesdosis im Bereich von 0,1 mg bis 100 mg (typischerweise von 0,1 mg und 50 mg) pro Tag pro Kilogramm Körpergewicht, z.B. 0,1-10 mg/kg/Tag. Tabletten oder Kapseln, können beispielsweise von 0,01 bis 100 mg, typischerweise von 0,02 bis 50 mg enthalten. Im Falle pharmazeutisch verträglicher Salze beziehen sich die vorgenannten Gewichtsangaben auf das Gewicht des vom Salz abgeleiteten Diphenylazetidinon-Ions. Zur Prophylaxe oder Therapie der oben genannten Zustände können die Verbindungen gemäß Formel (I) selbst als Verbindung verwendet werden, vorzugsweise liegen sie jedoch mit einem verträglichen Träger in Form einer pharmazeutischen Zusammensetzung vor. Der Träger muß natürlich verträglich sein, in dem Sinne, daß er mit den anderen Bestandteilen der Zusammensetzung kompatibel ist und nicht gesundheitsschädlich für den Patienten ist. Der Träger kann ein Feststoff oder eine Flüssigkeit oder beides sein und wird vorzugsweise mit der Verbindung als Einzeldosis formuliert, beispielsweise als Tablette, die von 0,05% bis 95 Gew.-% des Wirkstoffs enthalten kann. Weitere pharmazeutisch aktive Substanzen können ebenfalls vorhanden sein, einschließlich weiterer Verbindungen gemäß Formel (I). Die erfindungsgemäßen pharmazeutischen Zusammensetzungen können nach einer der bekannten pharmazeutischen Methoden hergestellt werden, die im wesentlichen darin bestehen, daß die Bestandteile mit pharmakologisch verträglichen Träger- und/oder Hilfsstoffen
gemischt werden.
Erfindungsgemäße pharmazeutische Zusammensetzungen sind solche, die für orale und perorale (z.B. sublinguale) Verabreichung geeignet sind, wenngleich die geeignetste Verabreichungsweise in jedem Einzelfall von der Art und Schwere des zu behandelnden Zustandes und von der Art der jeweils verwendeten Verbindung gemäß Formel (I) abhängig ist. Auch dragierte Formulierungen und dragierte Retardformulierungen gehören in den Rahmen der Erfindung. Bevorzugt sind säure- und magensaftresistente Formulierungen. Geeignete magensaftresistente Beschichtungen umfassen Celluloseacetatphthalat, Polyvinalacetatphthalat,
Hydroxypropylmethylcellulosephthalat und anionische Polymere von Methacrylsäure und Methacrylsäuremethylester.
Geeignete pharmazeutische Verbindungen für die orale Verabreichung können in separaten Einheiten vorliegen, wie zum Beispiel Kapseln, Oblatenkapseln,
Lutschtabletten oder Tabletten, die jeweils eine bestimmte Menge der Verbindung gemäß Formel (I) enthalten; als Pulver oder Granulate; als Lösung oder Suspension in einer wäßrigen oder nicht-wäßrigen Flüssigkeit; oder als eine Öl-in-Wasser- oder Wasser-in Öl-Emulsion. Diese Zusammensetzungen können, wie bereits erwähnt, nach jeder geeigneten pharmazeutischen Methode zubereitet werden, die einen
Schritt umfaßt, bei dem der Wirkstoff und der Träger (der aus einem oder mehreren zusätzlichen Bestandteilen bestehen kann) in Kontakt gebracht werden. Im allgemeinen werden die Zusammensetzungen durch gleichmäßiges und homogenes Vermischen des Wirkstoffs mit einem flüssigen und/oder feinverteilten festen Träger hergestellt, wonach das Produkt, falls erforderlich, geformt wird. So kann beispielsweise eine Tablette hergestellt werden, indem ein Pulver oder Granulat der Verbindung verpreßt oder geformt wird, gegebenenfalls mit einem oder mehreren zusätzlichen Bestandteilen. Gepreßte Tabletten können durch Tablettieren der Verbindung in frei fließender Form, wie beispielsweise einem Pulver oder Granulat, gegebenenfalls gemischt mit einem Bindemittel, Gleitmittel, inertem Verdünner und/oder einem (mehreren) oberflächenaktiven/dispergierenden Mittel in einer geeigneten Maschine hergestellt werden. Geformte Tabletten können durch Formen
der pulverförmigen, mit einem inerten flüssigen Verdünnungsmittel befeuchteten Verbindung in einer geeigneten Maschine hergestellt werden.
Pharmazeutische Zusammensetzungen, die für eine perorale (sublinguale) Verabreichung geeignet sind, umfassen Lutschtabletten, die eine Verbindung gemäß Formel (I) mit einem Geschmacksstoff enthalten, üblicherweise Saccharose und Gummi arabicum oder Tragant, und Pastillen, die die Verbindung in einer inerten Basis wie Gelatine und Glycerin oder Saccharose und Gummi arabicum umfassen.
Als weitere Wirkstoffe für die Kombinationspräparate sind geeignet:
Alle Antidiabetika, die in der Roten Liste 2005, Kapitel 12 genannt sind; alle Abmagerungsmittel/Appetitzügler, die in der Roten Liste 2005, Kapitel 1 gennant sind; alle Lipidsenker, die in der Roten Liste 2005, Kapitel 58 genannt sind. Sie können mit der erfindungsgemäßen Verbindung der Formel I insbesondere zur synergistischen Wirkungsverbesserung kombiniert werden. Die Verabreichung der
Wirkstoffkombination kann entweder durch getrennte Gabe der Wirkstoffe an den Patienten oder in Form von Kombinationspräparaten, worin mehrere Wirkstoffe in einer pharmazeutischen Zubereitung vorliegen, erfolgen. Die meisten der nachfolgend aufgeführten Wirkstoffe sind in USP Dictionary of USAN and International Drug Names, US Pharmacopeia, Rockville 2001 , offenbart.
Antidiabetika umfassen Insulin und Insulinderivate, wie z.B. Lantus® (siehe www.lantus.com) oder HMR 1964 oder solche, wie sie in WO2005005477 (Novo Nordisk) beschrieben sind, schnell wirkende Insuline (siehe US 6,221 ,633), inhalierbare Insuline, wie z. B. Exubera ® oder orale Insuline, wie z. B. IN-105 (Nobex) oder Oral-lyn ™ (Generex Biotechnology), GLP-1 -Derivate wie z.B. Exenatide,
Liraglutide oder diejenigen die in WO 98/08871 , WO2005027978 von Novo Nordisk A/S, in WO 01/04156 von Zealand oder in WO 00/34331 von Beaufour-Ipsen offenbart wurden, Pramlintide Acetat (Symlin; Amylin Pharmaceuticals), sowie oral wirksame hypoglykämische Wirkstoffe.
Die oral wirksamen hypoglykämischen Wirkstoffe umfassen vorzugsweise
Sulfonylharnstoffe,
Biguanidine,
Meglitinide,
Oxadiazolidindione, Thiazolidindione,
Glukosidase-Inhibitoren,
Hemmstoffe der Glykogenphosphorylase,
Glukagon-Antagonisten,
Glukokinaseaktivatoren, Inhibitoren der Fructose-1 ,6-bisphosphatase
Modulatoren des Glukosetransporters-4 (GLUT4),
Inhibitoren der Glutamin-Fructose-6-Phosphat-Amidotransferase (GFAT),
GLP-1-Agonisten, Kaliumkanalöffner, wie z.B. diejenigen, die in WO 97/26265 und
WO 99/03861 von Novo Nordisk A/S offenbart wurden, Inhibitoren der Dipeptidylpeptidase-IV (DPP-IV),
Insulin-Sensitizer,
Inhibitoren von Leberenzymen, die an der Stimulation der Glukoneogenese und/oder
Glykogenolyse beteiligt sind,
Modulatoren der Glukoseaufnahme, des Glukosetransports und der Glukoserückresorption,
Hemmstoffe der 11 ß-HSD1 ,
Inhibitoren der Protein-Tyrosin-Phosphatase-1 B (PTP1B),
Modulatoren des natrium-abhängigen Glukosetransporters 1 oder 2 (SGLT1 , SGLT2), den Fettstoffwechsel verändernde Verbindungen wie antihyperlipidämische Wirkstoffe und antilipidämische Wirkstoffe,
Verbindungen, die die Nahrungsmitteleinnahme verringern,
Verbindungen, die die Thermogenese erhöhen,
PPAR- und RXR-Modulatoren und
Wirkstoffe, die auf den ATP-abhängigen Kaliumkanal der Betazellen wirken.
Bei einer Ausführungsform der Erfindung wird die Verbindungen der Formel I in
Kombination mit einem HMGCoA-Reduktase Inhibitor wie Simvastatin, Fluvastatin,
Pravastatin, Lovastatin, Atorvastatin, Cerivastatin, Rosuvastatin, L-659699 verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Cholesterinresorptionsinhibitor, wie z.B. Ezetimibe, Tiqueside, Pamaqueside, FM-VP4 (sitostanol/campesterol ascorbyl phosphat; Forbes Medi-Tech, WO2005042692), MD-0727 (Microbia Inc., WO2005021497) oder mit Verbindungen, wie in WO2002066464 (Kotobuki Pharmaceutical Co. Ltd.) oder WO2005062824 (Merck & Co.) oder WO2005061451 und WO2005061452 (AstraZeneca AB) beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem PPAR gamma Agonisten, wie z.B. Rosiglitazon, Pioglitazon, JTT-501 , Gl 262570, R-483, CS-011 (Rivoglitazon) verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit PPAR alpha Agonist, wie z.B. GW9578, GW-590735, K-111 , LY-674, KRP-101 , DRF-10945 verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem gemischten PPAR alpha/gamma Agonisten, wie z.B. Muraglitazar, Tesaglitazar, Naveglitazar, LY-510929, ONO-5129, E-3030, AVE 8042, AVE 8134, AVE 0847, oder wie in PCT/US 00/11833, PCT/US 00/11490, DE10142734.4 oder in J.P.Berger et al., TRENDS in Pharmacological Sciences 28(5), 244-251 , 2005 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem PPAR delta Agonisten, wie z.B. GW-501516 verabreicht. Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Metaglidasen oder mit MBX-2044 oder anderen partiellen PPAR gamma Agonisten/Antagonisten verabreicht
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Fibrat, wie z.B. Fenofibrat, Clofibrat, Bezafibrat, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem MTP-Inhibitor, wie z.B. Implitapide , BMS-201038, R-103757 oder solchen wie in WO2005085226 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem CETP-Inhibitor, wie z.B. Torcetrapib oder JTT-705 , verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Gallensäureresorptionsinhibitor (siehe z.B. US 6,245,744, US 6,221 ,897 oder WO00/61568), wie z.B. HMR 1741 oder solchen wie in DE 10 2005 033099.1 und DE 10 2005 033100.9 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem polymeren Gallensäureadsorber, wie z.B. Cholestyramin, Colesevelam, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem LDL-Rezeptorinducer (siehe US 6,342,512), wie z.B. HMR1171 , HMR1586, oder solchen wie in WO2005097738 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Omacor® (Omega-3-Fettsäuren; hochkonzentrierte Ethylester der Eicosapentaensäure und der Docosahexaensäure) verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem ACAT-Inhibitor, wie z.B. Avasimibe, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in
Kombination mit einem Antioxidans, wie z.B. OPC-14117, Probucol, Tocopherol, Ascorbinsäure, ß-Caroten oder Selen verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Vitamin, wie z. B. Vitamin B6 oder Vitamin B12 verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Lipoprotein-Lipase Modulator, wie z.B. Ibrolipim (NO-1886), verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem ATP-Citrat-Lyase Inhibitor, wie z.B. SB-204990, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Squalen Synthetase Inhibitor, wie z.B. BMS-188494 oder wie in WO2005077907 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Lipoprotein(a) antagonist, wie z.B. Gemcabene (CI-1027) verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem HM74A Rezeptor Agonisten, wie z.B. Nicotinsäure, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Lipase Inhibitor, wie z.B. Orlistat oder Cetilistat (ATL-962), verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Insulin verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Sulfonylharnstoff, wie z.B. Tolbutamid, Glibenclamid, Glipizid oder Glimepirid verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Biguanid, wie z.B. Metformin, verabreicht.
Bei wieder einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Meglitinid, wie z.B. Repaglinide oder Nateglinid, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Thiazolidindion, wie z.B. Troglitazon, Ciglitazon, Pioglitazon, Rosiglitazon oder den in WO 97/41097 von Dr. Reddy's Research Foundation offenbarten Verbindungen, insbesondere 5-[[4-[(3,4-Dihydro-3-methyl-4-oxo-2-chinazolinylmethoxy]- phenyl]methyl]-2,4-thiazolidindion, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem α-Glukosidase-lnhibitor, wie z.B. Miglitol oder Acarbose, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Wirkstoff verabreicht, der auf den ATP-abhängigen Kaliumkanal der Betazellen wirkt, wie z.B. Tolbutamid, Glibenclamid, Glipizid, Glimepirid oder Repaglinid.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit mehr als einer der vorstehend genannten Verbindungen, z.B. in Kombination mit einem Sulfonylharnstoff und Metformin, einem Sulfonylharnstoff und Acarbose, Repaglinid und Metformin, Insulin und einem Sulfonylharnstoff, Insulin und Metformin, Insulin und Troglitazon, Insulin und Lovastatin, etc. verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Hemmstoff der Glykogenphosphorylase, wie z.B. PSN-357 oder FR-258900 oder solchen wie in WO2003084922, WO2004007455, WO2005073229-31 , WO2005067932 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Glukagon-Rezeptor-Antagonisten, wie z.B. A-770077 oder NNC-25-2504 oder wie in WO2004100875, WO2005065680, beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Aktivatoren der Glukokinase, wie z. B. LY-2121260 (WO2004063179), PSN-105, PSN-110, GKA-50 oder solchen wie sie z. B. in WO2004072031 oder WO2004072066 oder WO2005080360 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Glukoneogenese, wie z. B. FR-225654, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der Fructose-1 ,6-bisphosphatase (FBPase) wie z.B. CS-917, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Modulatoren des Glukosetransporters-4 (GLUT4), wie z. B. KST-48 (D.-O. Lee et al.: Arzneim.-Forsch. Drug Res. 54 (12), 835 (2004)), verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit
Inhibitoren der Glutamin-Fructose-6-Phosphat-Amidotransferase (GFAT), wie sie z. B. in WO2004101528 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der Dipeptidylpeptidase-IV (DPP-IV), wie z. B. Vildagliptin (LAF-237),
Sitagliptin (MK-0431), Saxagliptin ((BMS-477118), GSK-823093, PSN-9301 , SYR-322, SYR-619, TA-6666, TS-021 , GRC-8200, GW-825964X oder wie sie in WO2003074500, WO2003106456, WO200450658, WO2005058901 , WO2005012312, WO2005/012308, PCT/EP2005/007821 , PCT/EP2005/008005, PCT/EP2005/008002, PCT/EP2005/008004, PCT/EP2005/008283, DE 10 2005 012874.2 oder DE 10 2005 012873.4 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Hemmstoffen der 11-beta-Hydroxysteroid-Dehydrogenase-1 (11 ß-HSD1), wie z. B. BVT-2733 oder solche, wie sie z. B. in WO200190090-94, WO200343999, WO2004112782, WO200344000, WO200344009, WO2004112779, WO2004113310, WO2004103980, WO2004112784, WO2003065983, WO2003104207, WO2003104208, WO2004106294, WO2004011410, WO2004033427, WO2004041264, WO2004037251 , WO2004056744, WO2004065351 , WO2004089367, WO2004089380, WO2004089470-71 , WO2004089896, WO2005016877, WO2005097759 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der Protein-Tyrosin-Phosphatase-1 B (PTP1 B), wie sie z. B. in WO200119830-31 , WO200117516, WO2004506446, WO2005012295, PCT/EP2005/005311 , PCT/EP2005/005321 , PCT/EP2005/007151 , PCT/EP2005/ oder DE 10 2004 060542.4 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Modulatoren des natrium-abhängigen Glukosetransporters 1 oder 2 (SGLT1 , SGLT2), wie z.B. KGA-2727, T-1095, SGL-0010, AVE 2268 und SAR 7226 oder wie sie z. B. in WO2004007517, WO200452903, WO200452902, PCT/EP2005/005959,
WO2005085237, JP2004359630 oder von A. L. Handion in Expert Opin. Ther. Patents (2005) 15(11), 1531-1540 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Modulatoren des GPR40 verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der hormon-sensitiven Lipase (HSL), wie z. B. in WO2005073199 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Hemmstoffen der Acetyl-CoA Carboxylase (ACC) wie z. B. solchen wie in
W0199946262, WO200372197, WO2003072197, WO2005044814 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Phosphoenolpyruvatcarboxykinase (PEPCK), wie z.B. solchen, wie in WO2004074288 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Glykogen Synthase Kinase-3 beta (GSK-3 beta), wie z. B. in US2005222220, WO2005085230, PCT/EP2005/005346, WO2003078403, WO2004022544, WO2003106410, WO2005058908, US2005038023, WO2005009997, US2005026984, WO2005000836, WO2004106343, EP1460075, WO2004014910, WO2003076442, WO2005087727, WO2004046117 beschrieben.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Protein Kinase C beta (PKC beta), wie z. B. Ruboxistaurin, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Endothelin-A-Rezeptor Antagonisten, wie z. B. Avosentan (SPP-301), verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der „l-kappaB kinase" (IKK Inhibitoren), wie sie z. B. in WO2001000610, WO2001030774, WO2004022553, WO2005097129 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Modulatoren des Glucocorticoidrezeptors, wie sie z. B. in WO2005090336 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit CART-Modulatoren (siehe "Cocaine-amphetamine-regulated transcript influences energy metabolism, anxiety and gastric emptying in mice" Asakawa, A. et al.: Hormone and Metabolie Research (2001), 33(9), 554-558);
NPY-Antagonisten wie z.B. Naphthalin-1-sulfonsäure-{4-[(4-amino-quinazolin-2- ylamino)-methyl]-cyclohexylmethyl}-amid Hydrochlorid (CGP 71683A); Peptid YY 3-36 (PYY3-36) oder analoge Verbindungen wie z. B. CJC-1682 (PYY3-36 konjugiert mit humanem Serum Albumin über Cys34) oder CJC-1643 (Derivat des PYY3-36, welches sich in vivo an Serum Albumin konjugiert) oder solche, wie sie in WO2005080424 beschrieben sind;
Cannabinoid Rezeptor 1 Antagonisten (wie z.B. Rimonabant, SR147778 oder solche wie sie in z. B. EP 0656354, WO 00/15609, WO 02/076949, WO2005080345, WO2005080328, WO2005080343, WO2005075450, WO2005080357, WO200170700, WO2003026647-48, WO200302776, WO2003040107,
WO2003007887, WO2003027069, US6,509,367, WO200132663, WO2003086288, WO2003087037, WO2004048317, WO2004058145, WO2003084930, WO2003084943, WO2004058744, WO2004013120, WO2004029204, WO2004035566, WO2004058249, WO2004058255, WO2004058727, WO2004069838, US20040214837, US20040214855, US20040214856, WO2004096209, WO2004096763, WO2004096794, WO2005000809, WO2004099157, US20040266845, WO2004110453, WO2004108728, WO2004000817, WO2005000820, US20050009870, WO200500974, WO2004111033-34, WO200411038-39, WO2005016286, WO2005007111 , WO2005007628, US20050054679, WO2005027837, WO2005028456,
WO2005063761-62, WO2005061509, WO2005077897 beschrieben sind); MC4-Agonisten (z.B. 1-Amino-1 ,2,3,4-tetrahydro-naphthalin-2-carbonsäure [2-(3a- benzyl-2-methyl-3-oxo-2,3,3a,4,6,7-hexahydro-pyrazolo[4,3-c]pyridin-5-yl)-1-(4-chloro- phenyl)-2-oxo-ethyl]-amid; (WO 01/91752)) oder LB53280, LB53279, LB53278 oder THIQ, MB243, RY764, CHIR-785, PT-141 oder solche wie sie in WO2005060985, WO2005009950, WO2004087159, WO2004078717, WO2004078716, WO2004024720, US20050124652, WO2005051391 , WO2004112793, WOUS20050222014, US20050176728, US20050164914, US20050124636, US20050130988, US20040167201 , WO2004005324, WO2004037797, WO2005042516, WO2005040109, WO2005030797, US20040224901 ,
WO200501921 , WO200509184, WO2005000339, EP1460069, WO2005047253, WO2005047251 , EP1538159, WO2004072076, WO2004072077 beschrieben sind;
Orexin-Rezeptor Antagonisten (z.B. 1-(2-Methyl-benzoxazol-6-yl)-3-[1 ,5]naphthyridin- 4-yl-harnstoff Hydrochlorid (SB-334867-A) oder solche, wie sie z. B. in WO200196302, WO200185693, WO2004085403, WO2005075458 beschrieben sind); Histamin H3 Rezeptor Agonisten (z. B. 3-Cyclohexyl-1-(4,4-dimethyl-1 ,4,6,7- tetrahydro-imidazo[4,5-c]pyridin-5-yl)-propan-1-on Oxalsäuresalz (WO 00/63208) oder solche, wie sie in WO200064884, WO2005082893 beschrieben sind); CRF-Antagonisten (z.B. [2-Methyl-9-(2,4,6-trimethyl-phenyl)-9H-1 ,3,9-triaza-fluoren-4- yl]-dipropyl-amin (WO 00/66585)); CRF BP-Antagonisten (z.B. Urocortin); Urocortin-Agonisten; ß3-Agonisten (wie z.B. 1-(4-Chloro-3-methanesulfonylmethyl-phenyl)-2-[2-(2,3- dimethyl-1 H-indol-6-yloxy)-ethylamino]-ethanol Hydrochlorid (WO 01/83451)); MSH (Melanocyt-stimulierendes Hormon)-Agonisten; MCH (melanin-konzentrierendes Hormon) Rezeptor Antagonisten (wie z. B. NBI-845, A-761 , A-665798, A-798, ATC-0175, T-226296, T-71 , GW-803430 oder solche Verbindungen, wie sie in WO2003/15769, WO2005085200, WO2005019240, WO2004011438, WO2004012648, WO2003015769, WO2004072025, WO2005070898, WO2005070925, WO2004039780, WO2003033476, WO2002006245, WO2002002744, WO2003004027, FR2868780 beschrieben sind); CCK-A Agonisten (wie z.B. {2-[4-(4-Chloro-2,5-dimethoxy-phenyl)-5-(2-cyclohexyl- ethyl)-thiazol-2-ylcarbamoyl]-5,7-dimethyl-indol-1-yl}-essigsäure Trifluoressigsäuresalz (WO 99/15525) oder SR-146131 (WO 0244150) oder SSR-125180); Serotonin-Wiederaufnahme-Inhibitoren (z.B. Dexfenfluramine); gemischte Sertonin- und noradrenerge Verbindungen (z.B. WO 00/71549);
5-HT-Rezeptor Agonisten z.B. 1-(3-Ethyl-benzofuran-7-yl)-piperazin Oxalsäuresalz (WO 01/09111);
5-HT2C Rezeptor Agonisten (wie z.B. APD-356 oder BVT-933 oder solche, wie sie in WO200077010, WO20077001-02, WO2005019180, WO2003064423, WO200242304, WO2005082859 beschrieben sind);
5-HT6 Rezeptor Antagonisten, wie sie z.B. in WO2005058858 beschrieben sind; Bombesin-Rezeptor Agonisten (BRS-3 Agonisten;
Galanin-Rezeptor Antagonisten;
Wachstumshormon (z.B. humanes Wachstumshormon oder AOD-9604);
Wachstumshormon freisetzende Verbindungen (6-Benzyloxy-1-(2-diisopropylamino- ethylcarbamoyO-S^-dihydro-I H-isochinolin^-carbonsäuretertiärbutylester (WO 01/85695));
Growth Hormone Secretagogue Receptor Antagonisten (Ghrelin Antagonisten) wie z.
B. A-778193 oder solchen, wie sie in WO2005030734 beschrieben sind;
TRH-Agonisten (siehe z.B. EP 0 462 884); entkoppelnde Protein 2- oder 3-Modulatoren; Leptinagonisten (siehe z.B. Lee, Daniel W.; Leinung, Matthew C; Rozhavskaya-
Arena, Marina; Grasso, Patricia. Leptin agonists as a potential approach to the treatment of obesity. Drugs of the Future (2001), 26(9), 873-881);
DA-Agonisten (Bromocriptin, Doprexin);
Lipase/Amylase-Inhibitoren (z.B. WO 00/40569); Inhibitoren der Diacylglycerol O-Acyltransferasen (DGATs) wie z. B. in
US2004/0224997, WO2004094618, WO200058491 , WO2005044250,
WO2005072740, JP2005206492, WO2005013907 beschrieben;
Inhibitoren der Fettsäuresynthase (FAS) wie z.B. C75 oder solchen, wie in
WO2004005277 beschrieben; Oxyntomodulin;
Oleoyl-Estron
oder Agonisten des Schilddrüsenhormonrezeptors (thyroid hormone receptor agonists) wie z. B: KB-2115 oder solche, wie in WO20058279, WO200172692, WO200194293, WO2003084915, WO2004018421 , WO2005092316 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung ist der weitere Wirkstoff Leptin; siehe z.B. "Perspectives in the therapeutic use of leptin", Salvador, Javier; Gomez- Ambrosi, Javier; Fruhbeck, Gema, Expert Opinion on Pharmacotherapy (2001), 2(10), 1615-1622.
Bei einer Ausführungsform ist der weitere Wirkstoff Dexamphetamin oder Amphetamin.
Bei einer Ausführungsform ist der weitere Wirkstoff Fenfluramin oder Dexfenfluramin.
Bei noch einer Ausführungsform ist der weitere Wirkstoff Sibutramin.
Bei einer Ausführungsform ist der weitere Wirkstoff Mazindol oder Phentermin.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Ballaststoffen, vorzugsweise unlöslichen Ballaststoffen (siehe z.B. Carob/ Caromax® (Zunft H J; et al., Carob pulp preparation for treatment of hypercholesterolemia, ADVANCES IN THERAPY (2001 Sep-Oct), 18(5), 230-6.) Caromax ist ein Carob enthaltendes Produkt der Fa. Nutrinova, Nutrition Specialties &Food Ingredients GmbH, Industriepark Höchst, 65926 Frankfurt / Main)) verabreicht. Die Kombination mit Caromax® kann in einer Zubereitung erfolgen, oder durch getrennte Gabe von Verbindungen der Formel I und Caromax®. Caromax® kann dabei auch in Form von Lebensmitteln, wie z.B. in Backwaren oder Müsliriegeln, verabreicht werden.
Es versteht sich, dass jede geeignete Kombination der erfindungsgemäßen Verbindungen mit einer oder mehreren der vorstehend genannten Verbindungen und wahlweise einer oder mehreren weiteren pharmakologisch wirksamen Substanzen als unter den Schutzbereich der vorliegenden Erfindung fallend angesehen wird.
CH2-CH3



LY-674 KRP-101

LY-510929 GW-501516





Bevorzugt sind racemische als auch enantiomerenreine Verbindungen der Formel I mit folgender Struktur:
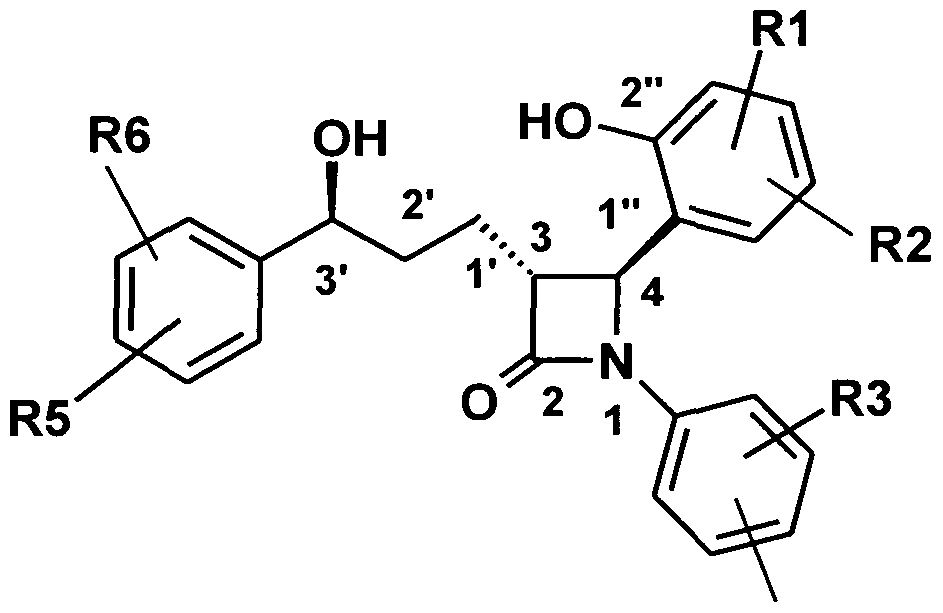
R4
Als Aminoschutzgruppen werden bevorzugt der durch katalytische Hydrierung abspaltbare Benzyloxycarbonyl-(Z-)Rest, der durch schwache Säuren abspaltbare 2- (3,5-Dimethyloxyphenyl)propyl(2)oxycarbonyl (Ddz-) oder Trityl- (Trt)-Rest, der durch Säuren wie 3M Salzsäure abspaltbare t-Butylcarbamat (BOC-)-Rest und der durch sekundäre Amine abspaltbare 9-Fluorenylmethyloxycarbonyl- (Fmoc)-Rest herangezogen.
Die Erfindung betrifft weiterhin ein Verfahren zur Herstellung von Diphenylazetidinonderivaten der Formel I.


Y, W, Z, Y', W, Z1 können unabhängig voneinander -NH-, -O-, -(C=O), Phenyl, Cycloalkyl, Heterocycloalkyl oder eine Bindung bedeuten und LAG kann die Bedeutungen wie weiter vorn beschrieben haben.
Die Verknüpfung von -(CH2)o-i-Y-W-Z-(Co-C25)-Alkylen-H in Verbindung Il kann alternativ auch an einem der anderen beiden Phenylringen sein.
Das Verfahren zur Herstellung der Verbindungen der Formel I ist dadurch gekennzeichnet, daß man z.B. ein aktiviertes in geeigneter Weise geschütztes
Carbamat oder ein in geeigneter Weise geschütztes alpha, omega-Diol oder einen in
geeigneter Weise geschützten alpha, omega-Halogenoalkohol oder ein Alkylierungsmittel mit einem Amin oder einer Hydroxy-Verbindung der Formel II, wobei die Hydroxyfunktionen in 3'- bzw 2"-Stellung in geeigneter Weise geschützt sein können, umsetzt. Nach Abspaltung der eventuell vorhandenen Schutzgruppe kann die Verbindunge der Formel Il in einem weiteren Schritt mit der Gruppe LAG verknüpft werden, z.B. unter Ausbildung von Schwefelsäuremonoamiden.
Die nachfolgenden Beispiele dienen zur näheren Erläuterung der Erfindung, ohne dieselbe auf in den Beispielen beschriebene Produkte und Ausführungsformen einzuschränken.
Schema 1

,2,7-Trimethyl-benzoM .31dioxin-4-on 2

7-Bromomethyl-212-dimethyl-benzori ,31dioxin-4-on 3
100 g (0.52 mol) Acetonid 2 werden in 1 I Tetrachlorkohlenstoff gelöst. Nach Zugabe von 6 g Benzoylperoxid wird 2 Stunden am Rückfluß gekocht. Danach kühlt man auf Raumtemperatur ab und verdünnt mit 500 ml n-Heptan/Ethylacetat (3:1). Diese Lösung wird über Kieselgel filtriert und nicht ganz zur Trockene eingeengt (rund 50 ml Lösungsmittelrückstand). Diese Lösung wird bei -300C über Nacht gelagert. Das ausgefallene Produkt wird abgesaugt und man erhält 69.5 g kristallines Benzylbromid 3, das noch mit wenig Peroxyd verunreinigt ist.
Essiqsäure-2,2-dimethyl-4-oxo-4H-benzoM ,31dioxin-7-ylmethyl ester 4

55 g (0.20 mol) Benzylbromid 3 werden in 550 ml DMF gelöst. Nach Zugabe von 30 g (.0.37 mol) Natriumacetat lässt man über Nacht stehen. Zur Aufarbeitung wird mit 700 ml n-Heptan/Ethylacetat (2:1) und 500 ml Wasser versetzt. Die wässrige Phase wird abgetrennt, die organische Phase 2 mal mit wässriger NaCI-Lösung gewaschen, über Kieselgel filtriert und eingeengt. Der Rückstand wird mit Flashchromatographie (n-
Heptan/Ethylacetat 4:1 bis 2:1) gereinigt und man erhält 22.9 g amorphes Produkt 4.
2-Hvdroxy-4-hvdroxymethyl-benzosäure-methylester 5

30 g (0.12 mol) Verbindung 4 werden in 100 ml Methanol suspendiert. Unter Rühren werden 300 ml 1 M NaOMe/MeOH Lösung zugetropft, wobei kurzzeitig eine klare Lösung entsteht. Nach 30 Minuten rühren wird die Lösung mit 320 ml 1 M HCI/MeOH sauer gestellt (die Lösung wird dabei farblos) und über wenig Kieselgel das entstandene NaCI abgetrennt. Dann wird eingeengt und der Rückstand in 100 ml n- Heptan/Ethylacetat (2:1) suspendiert. Diese Lösung wird erneut über Kieselgel filtriert und mit der n-Heptan/Ethylacetat-Mischung nachgewaschen. Nach dem Abrauchen des Lösungsmittels erhält man 18.1 g Rohprodukt 5.
2-Benzyloxy-4-benzyloxymethyl-benzosäure-methylester β

Rückstand mit Flashchromatographie (n-Heptan/Ethylacetat 6:1 bis 2:1) gereinigt. Man erhält 21.4 g perbenzyliertes Produkt 6.
(2-Benzyloxy-4-benzyloxymethyl-phenyl)-methanol 7

21.4 g (59.2 mmol) Methylester 6 werden in 270 ml THF gelöst und auf O0C gekühlt. Es wird eine 1 M Lithiumaluminiumhydrid-Lösung in Diethylether bei O0C langsam zugetropft und anschließend 15 Minuten bei 00C gerührt. Überschüssiges Lithiumaluminiumhydrid wird durch Zugabe von 3 ml Ethylacetat zerstört. Um einen gut filtrierbaren Niederschlag zu bekommen, werden nacheinander vorsichtig 4 ml Wasser, 4 ml 10 %-ige Natronlauge und 8 ml Wasser zugegeben. Der Niederschlag wird über Kieselgel filtriert, mit Ethylacetat nachgewaschen und dann eingeengt. Man erhält 19.7 g Rohprodukt 7.
2-Benzyloxy-4-benzyloxymethyl-benzaldehvd 8

19.7 g Rohprodukt 7 werden in 300 ml DMSO und 150 ml Essigsäureanhydrid gelöst und 18 Stunden bei Raumtemperatur stehen gelassen. Diese Reaktionslösung wird dann mit 500 ml n-Heptan/Ethylacetat verdünnt und 3 mal mit gesättigter NaCI- Lösung gewaschen, über Kieselgel filtriert und eingeengt. Restliches Essigsäureanhydrid wird mit Toluol abgeraucht und man erhält 19.2 g Aldehyd 8 als Rohprodukt.
(2-Benzyloxy-4-benzyloxymethyl-benzylidene)-(4-fluoro-phenyl)-amin 10

19.2 g Aldehyd 8 und 10 ml (103 mmol) p-Fluoranilin 9 (Fluka) werden mit 300 ml Toluol 2 Stunden am Wasserabscheider gekocht und man destilliert dabei rund 100 ml Toluol ab. Das restliche Toluol wird am Rotationsverdampfer eingeengt und der Rückstand wird mit Flashchromatographie (n-Heptan/Ethylacetat 2:1 + 1% Triethylamin) gereinigt und man erhält 25 g Imin 10.
3-r2-r(2-Benzyloxy-4-benzyloxymethyl-phenyl)-(4-fluoro-phenylamino)-methyll-5-(tert- butyl-dimethyl-silanyloxy)-5-(4-fluoro-phenyl)-pentanovn-4-phenyl-oxazolidin-2-on 12

5.0 g (10.6 mmol) Oxazolidinon 11 werden zusammen mit 2.1 ml Diisopropylethylamin in 50 ml Methylenchlorid gelöst und unter Argon auf 00C gekühlt. Zu dieser Lösung tropft man langsam 8.8 ml einer 1 M TiCU/Methylenchlorid-Lösung zu. Dann wird 5 Minuten auf 200C erwärmt und danach auf -30°C abgekühlt. Bei -300C wird eine Lösung von 5.4 g (12.7 mmnol) Imin 10 in 15 ml Methylenchlorid zugetropft und 30 Minuten bei -300C gerührt. Die Reaktionslösung wird mit 100 ml Wasser extrahiert. Die organische Phase wird über 100 ml Kieselgel filtriert. Die wässrige Phase wird nochmals mit 80 ml n-Heptan/Ethylacetat (2:1) extrahiert und die organische Phase wird verwendet um das Kieselgel der ersten Filtration nachzuwaschen. Die organische Phase wird eingeengt und mit Flashchromatographie (n-Heptan/Ethylacetat 4 :1 bis
2:1 ) gereinigt. Man erhält 4.34 g Produkt 12.
4-(2-Benzyloxy-4-benzyloxymethyl-phenyl)-3-r3-(tert-butyl-dimethyl-silanyloxy)-3-(4- fluoro-phenyl)-propyll-1 -(4-fluoro-phenyl)-azetidin-2-on 13

4.34 g (4.8 mmol) Verbindung 12 werden in 60 ml Toluol gelöst. Man tropft 3.4 ml Bis- trimethylsilylacetamid (BSA) zu und kühlt auf 00C ab. Nach Zugabe von 1.7 ml 1 M Tetrabuylammoniumfluorid (TBAF) in THF lässt man auf Raumtemperatur erwärmen und rührt noch 1 Stunde bei Raumtemperatur nach. Die Reaktionslösung wird über Kieselgel filtriert und mit Ethylacetat nachgewaschen. Nach Abdestillieren des Lösungsmittels wird der Rückstand mit Flashchromatographie (n-Heptan/Ethylacetat 4:1 bis 2:1) gereinigt und man erhält 2.27 g beta-Lactan 13.
1-(4-Fluoro-phenyl)-3-f3-(4-fluoro-phenyl)-3-hvdroxy-propyn-4-(2-hydroxy-4- hydroxymethyl- phenyl)-azetidin-2-on 14
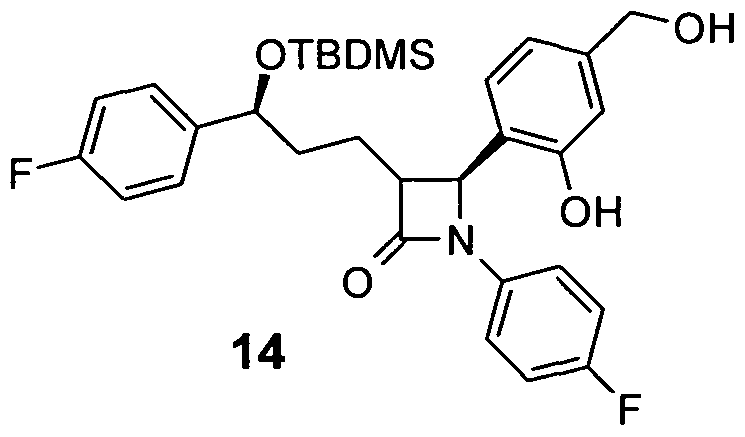
O

690 mg (1.25 mmol) Verbindung 14 werden in 10 ml Acetonitril gelöst. Man gibt nach einander 0.5 ml Triethylamin und 500 mg (1.9 mmol) Di-Su-CO (Fluka) zu und lässt 30 Minuten bei Raumtemperatur stehen. Dann tropft man die Reaktionslösung in eine Lösung von 1 g Piperazin in 10 ml Acetonitril und rührt 1 Stunde nach. Die heterogene Reaktionslösung wird direkt mit Flashchromatographie
(Methylenchlorid/Methanol/conz. Ammoniak 100/7/1 dann 30/5/1 dann 30/10/3) gereinigt und man erhält 590 mg Produkt 15 als farblosen amorphen Feststoff.
Piperazin-1-kohlensäure-4-(1-(4-fluoro-phenyl)-3-r3-(4-fluoro-phenyl)-3-hvdroxy- propyn-4-oxo-azetidin-2-yl)-3-hvdroxy-benzyl ester 16

580 mg (0.87 mmol) Verbindung 15 löst man in 10 ml THF. Nach Zugabe von 5 ml 2 N wässriger HCl lässt man die homogene Lösung 16 Stunden bei Raumtemperatur stehen. Die Lösung wird danach durch Zugabe einer Mischung von
Methylenchlorid/Methanol/conz. Ammoniak (30/10/30) basisch eingestellt und danach eingeengt. Der Rückstand wird in wenig 30/5/1 suspendiert und mit Flashchromatographie (Methylenchlorid/Methanol/conz. Ammoniak 30/5/1 dann 30/10/3) gereinigt und man erhält 400 mg Verbindung 16 als amorphen Feststoff mit dem Molekulargewicht 551.60 (C30H3IF2N3O5); MS (ESI+): 552.24 (M+H+).
4-(4-(1-(4-Fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-3-hvdroxy-propyn-4-oxo-azetidin-2-yl)- 3- hvdroxy-benzyloxycarbonyl)-piperazin-1-schwefelsäureamid Ammonium Salz 17 (Beispiel A)

560 mg (1.0 mmol) Verbindung 16 werden in 15 ml Methanol gelöst und auf 00C abgekühlt. Nach Zugabe von 1 g Trimethylaminschwefeltrioxid-Komplex rührt man 2 Stunden bei 00C. Die Reaktion wird durch Zugabe von 10 ml
Methylenchlorid/Methanol/conz. Ammoniak 30/10/3 abgebrochen und die Suspension über wenig Kieselgel filtriert und mit Methylenchlorid/Methanol/conz. Ammoniak 30/10/3 nachgewaschen. Dann wird eingeengt und der Rückstand mit Flashchromatographie (Methylenchlorid/Methanol/conz. Ammoniak 30/5/1 dann 30/10/3 dann 30/15/5) gereinigt. Man erhält 580 mg Produkt 17 als amorphen Feststoff mit dem Molekulargewicht 631.20 (C30H31F2N3O8S); MS (ESI"): 629.79 (M-H"
4-(4-Bromomethyl-2-hvdroxy-phenyl)-3-[3-(tert-butyl-dimethyl-silanyloxy)-3-(4-fluoro- phenvD-propyli-i -(4-fluoro-phenyl)-azetidin-2-on 18

100 mg (0.18 mmol) Benzylalkohol 14 werden in 3 ml DMF gelöst und auf -4O0C gekühlt. Es werden 180 mg Triphenylphosphindibromid (Aldrich) zugegeben und 20 Minuten bei -400C gerührt. Die Reaktionslösung wird mit 20 ml Ethylacetat verdünnt und 3 mal mit wässriger NaCI-Lösung gewaschen über wenig Kieselgel filtriert, eingeengt und man erhält 100 mg Rohprodukt. Dieses Rohprodukt wird in 20 ml THF gelöst und mit 10 ml 2 N wässriger HCl versetzt. Nach 18 Stunden bei Raumtemperatur wird mit 50 ml Ethylacetat verdünnt und nach Abtrennen der wässrigen Phase 2 mal mit Natriumhydrogencarbonat-Lösung gewaschen, über wenig Kieselgel filtriert, eingeengt und der Rückstand mit Flashchromatographie (n- Heptan/Ethylacetat 2:1 bis 1 :2) gereinigt. Man erhält 50 mg Benzylbromid 18.
1-(4-Fluoro-phenyl)-3-f3-(4-fluoro-phenyl)-3-hvdroxy-propyll-4-(2-hydroxy-4-piperazin- 1- ylmethyl-phenyl)-azetidin-2-on 19

40 mg (0.08 mmol) Bromid 18 werden in 2 ml Acetonitril gelöst. Nach Zugabe von 50 mg Piperazin rührt man 1 Stunde bei Raumtemperatur. Die Suspension wird
eingeengt und der Rückstand mit Flashchromatographie
(Methylenchlorid/Methanol/conz. Ammonik 30/5/1 dann 30/10/3) gereinigt. Man erhält 42 mg Amin 19.
4-(4-(1-(4-Fluoro-phenyl)-3-f3-(4-fluoro-phenyl)-3-hvdroxy-propyn-4-oxo-azetidin-2-yl}- 3- hydroxy-benzyl)-piperazin-1--schwefelsäureamid ammonium salz (Beispiel B)

4-Methyl-piperazin-1-kohlensäure-4-{1-(4-fluoro-phenyl)-3-r3-(4-fluoro-phenyl)-3- hvdroxy-propyll-4-oxo-azetidin-2-yl)-3-hvdroxy-benzyl ester 20

200 mg (0.36 mmol) Benzylalkohol 14 werden analog der Synthese von Verbindung 15 und 16 umgesetzt und man erhält 90 mg Methylpiperazin 20 als amorphen Feststoff.
4-(4-(1-(4-Fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-3-hvdroxy-propyll-4-oxo-azetidin-2-yl)- 3-hvdroxy-benzyloxycarbonyl)-1.1 -dimethyl-piperazin-1 -ium-iodid (Beispiel C)

80 mg (0.14 mmol) Verbindung 20 werden in 5 ml Toluol gelöst und auf 800C erwärmt. Zu dieser Lösung tropft man 0.8 ml Methyliodid zu und rührt 1 Stunde bei 800C. Das ausgefallene Produkt wird filtriert, mit Toluol nachgewaschen und man erhält 72 mg farblosen Feststoff des Amoniumsalzes Bsp. C mit dem Molekulargewicht 580.26 (C32H36F2N3O5); MS (ESI): 580.12 (M+).
4-(4-(1-(4-Fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-3-hvdroxy-propyll-4-oxo-azetidin-2-yl)- 3- hvdroxy-benzyloxycarbonylmethvD-i-methyl-pyridinium-iodid (Beispiel D)

135 mg (0.24 mmol) Benzylalkohol 14 werden analog der Synthese von Verbindung Beispiel C umgesetzt und man erhält 53 mg Ammoniumsalz Beispiel D als farblosen Feststoff mit dem Molekulargewicht 588.23 (C33H32F2N3O5); MS (ESI): 588.36 (M+).
Schema 2

DMSOZAc2O


75 g (410 mmol) 4-Nitro-salicylsäure (Aldrich) werden in 1 I DMF und 190 ml Benzylbromid gelöst. Nach Zugabe von 200 g K2CO3 wird die Suspension 16 Stunden bei Raumtemperatur kräftig gerührt. Die Reaktionslösung wird mit 1 I Ethylacetat und 1 I Wasser 15 Minuten gerührt. Die wässrige Phase wird abgetrennt und die organische Phase noch 2 mal mit gesättigter NaCI-Lösung gewaschen, über Kieselgel filtriert und eingeengt bis das Produkt anfängt auszukristallisieren. Die Kristalle werden abgesaugt (70g) und mit n-Heptan/Ethylacetat (4:1) gewaschen. Beim Einengen der Mutterlauge fällt eine weitere Kristallfraktion aus (dies kann noch 2 mal wiederholt werden). Insgesamt erhält man 132 g kristallines Produkt 21.
2-Benzyloxy-4-nitro-benzaldehyd 23

132 g (363 mmol) Nitroaromat 21 werden analog der Synthese von Verbindung 7 und 8 umgesetzt und man erhält Aldehyd 23 als farblosen Feststoff.
(2-Benzyloxy-4-nitro-benzylidene)-(4-fluoro-phenyl)-amin 24

4-(2-Benzyloxy-4-nitro-phenyl)-3-[3-(tert-butyl-dimethyl-silanyloxy)-3-(4-fluoro- phenyl)- propyli-1 -(4-fluoro-phenyl)-azetidin-2-on 26

Aus 27.5 g (78.5 mmol) Imin 24 und 62.2 g (132 mmol) Oxazolidinon 11 erhält man, bei analogem Herstellungsverfahren wie für Verbindung 12 und 13, 24 g beta-Lactam 26 als amorphen Feststoff.
4-(4-Amino-2-benzyloxy-phenyl)-3-r3-(tert-butyl-dimethyl-silanyloxy)-3-(4-fluoro- phenvD-propylM -(4-fluoro-phenyl)-azetidin-2-on 27
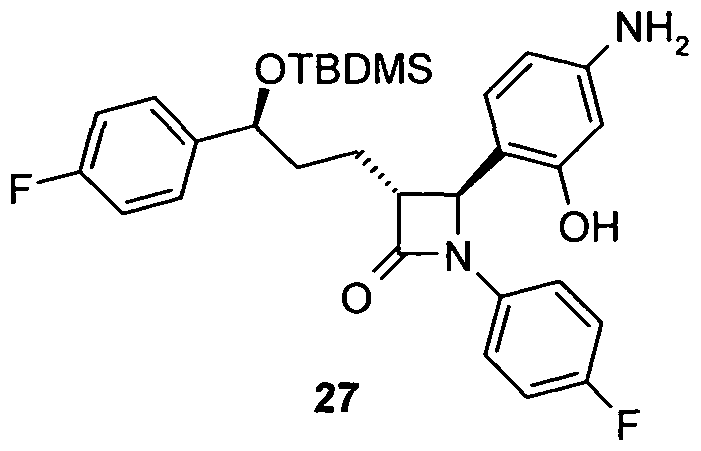
24 g (36 mmol) 26 werden analog der Hydrierung von Verbindung 13 umgesetzt und man erhält 13.2 g Anilin 27 als amorphen Feststoff.
Piperazin-1-kohlensäure-(4-[3-[3-(tert-butyl-dimethyl-silanyloxy)-3-(4-fluoro- phenvD- propvl1-1-(4-fluoro-phenvl)-4-oxo-azetidin-2-vll-3-hvdroxv-phenyl)-amid 28
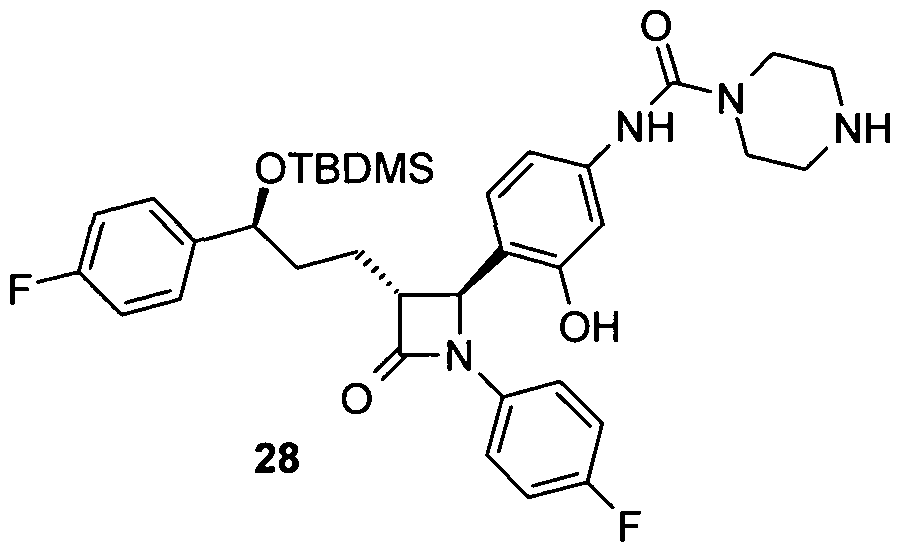
700 mg (1.3 mmol) Anilin 27 werden analog der Synthese von Amin 15 umgesetzt und man erhält 347 mg 28 als amorphen Feststoff.
Piperazine-1-kohlensäure-(4-{1-(4-fluoro-phenyl)-3-r3-(4-fluoro-phenyl)-3-hvdroxy- propyn-4-oxo-azetidin-2-yl)-3-hvdroxy-phenyl)-amid 29

4-(4-(1-(4-Fluoro-phenyl)-3-r3-(4-fluoro-phenyl)-3-hvdroxy-propyll-4-oxo-azetidin-2-ylV 3- hvdroxy-phenylcarbamoyl)-piperazin-1-schwefelsäureamid ammoniumsalz 30 (Beispiel E)

370 mg (0.69 mmol) Verbindung 29 werden analog der Synthese von Beispiel A umgesetzt und man erhält 343 mg Schwefelsäureamid 30 (Beispiel E) als amorphen Feststoff mit dem Molekulargewicht 616.18 (C29H30F2N4O7S); MS (ESI+): 1233.28 (2M+H+).
4-(4-Amino-2-hvdroxy-phenyl)-1-(4-fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-3-hvdroxy- propyll- azetidin-2-on 31

2.5 g (3.8 mmol) Verbindung 27 werden analog der Synthese von Verbindung 16 silylentschützt und man erhält 1.23 g Anilin 31 als amorphen Feststoff.
(4-(1-(4-Fluoro-phenyl)-3-r3-(4-fluoro-phenyl)-3-hydroxy-propyn-4-oxo-azetidin-2-yl)-3- hydroxy-phenyl)- schwefelsäureamid ammoniumsalz (Beispiel F)

44 mg (0.10 mmol) Anilin 31 werden analog der Synthese von Verbindung 30 mit 120 mg Trimethylamin-schwefeldioxid-Komplex sulfatiert und man erhält 21 mg Schwefelsäureamid Bsp. F als amorpher Feststoff mit dem Molekulargewicht 504.12 (C24H22F2N2O6S); MS (ESI"): 503.25 (M-H").
2,3,4,5,6-Pentahvdroxy-hexanoic acid (4-(1-(4-fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-3- hvdroxy-propyπ-4-oxo-azetidin-2-yl}-3-hvdroxy-phenyl)-amid (Beispiel G)

50 mg (0.12 mmol) Anilin 31 und 100 mg (0.24 mmol) Penta-O-acetylgluconsäure werden in 3 ml DMF gelöst. Man gibt 150 mg EDC und 75 mg HOBt zu und rührt 1 Stunde bei Raumtemperatur. Nach Zugabe von 20 ml Ethylacetat wird 3 mal mit NaCI- Lösung gewaschen über Kieselgel flitriert, eingeengt und der Rückstand mittels Flashchromatographie (n-Heptan/Ethylacetat 1 :1 bis 0:1) gereinigt. Das erhaltene Produkt (65 mg) wird in 5 ml Methanol gelöst und mit 0.5 ml 1 M NaOMe/MeOH versetzt. Nach 30 Minuten bei Raumtemperatur wird mit 0.5 M HCI/MeOH neutralisiert, eingeengt und der Rückstand mit Flashchromatographie
(Methylenchlorid/Methanol/conz. Ammoniak 30/5/1 dann 30/10/3 dann 30/15/5) gesäult. Man erhält 40 mg Zuckerderivat Bsp. G als amorphen Feststoff mit dem Molekulargewicht 602.59 (C30H32F2N2O9); MS (ESI'): 601.40 (M-H').
1-r(4-(1-(4-Fluoro-phenyl)-3-r3-(4-fluoro-phenyl)-3-hydroxy-propyn-4-oxo-azetidin-2-yl)- 3-hvdroxy-phenylcarbamoyl)-methyll-4-aza-1-azonia-bicvclor2.2.2loctan Jodid (Beispiel
H)

Bsp. H 54 mg (0.13 mmol) Anilin 31 und 150 mg Jodessigsäure werden analog mit EDC/HOBt gekoppelt wie bei der Synthese von Bsp. G beschrieben und man erhält 40 mg lodid. Dieses wird in 5 ml Toluol und 1 ml Methylenchlorid gelöst. Nach Zugabe von 200 ml DABCO wird 1 Stunde bei 800C gerührt. Der Niederschlag wird abgesaugt und man erhält das Trialkylammoniumalkysalz Bsp. H mit dem Molekulargewicht 577.26 (C32H35F2N4O4); MS (ESI): 577.20 (M+).
Schema 3



I1 R=SO3NH4*
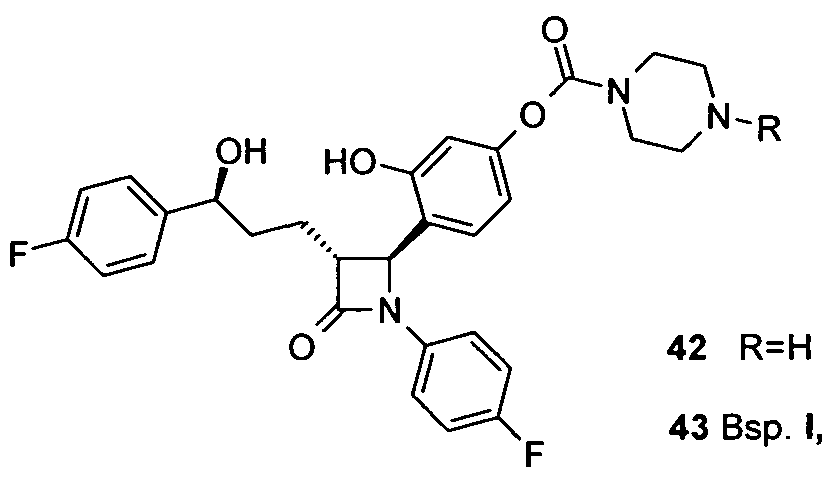 2,4-Dibenzyloxy-benzaldehvd 33
2,4-Dibenzyloxy-benzaldehvd 33

50 g (362 mmol) 2,4-Dihydroxy-benzaldehyd 32 (Aldrich) in 800 ml DMF mit 100 ml Benzylbromid und 200 g K2CO3 werden analog der Synthese von 21 perbenzyliert und man erhält 96 g kristallines Produkt 33.
(2,4-Dibenzyloxy-benzylidene)-(4-fluoro-phenyl)-amin 34

6.1 g (1.9 mmol) Aldehyd 33 wird mit 3.0 g (2.7 mmol) 4-Fluoranilin 9 analog der Synthese von Imin 10 umgesetzt und man erhält 6.0 g kristallines Imin 34.
3-r2-r(214-Dibenzyloxy-phenyl)-(4-fluoro-phenylamino)-methyll-5-(tert-butyl-dimethyl- silanyloxy)-5-(4-fluoro-phenyl)-pentanoyll-4-phenyl-oxazolidin-2-on 35

3-[3-(tert-Butyl-dimethyl-silanyloxy)-3-(4-fluoro-phenyl)-propyll-4-(2.4-dihvdroxy- phenyl)-1-(4-fluoro-phenyl)-azetidin-2-on 36
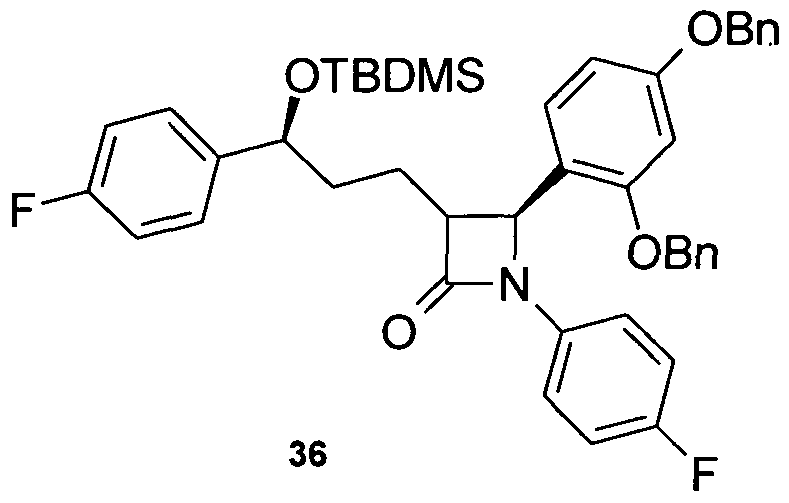
28.4 g (32.2 mmol) 35 werden analog der Synthese von beta-Lactam 13 cyclisiert und man erhält 13.1 g Lactam 36 als amorphen Feststoff.
4-(2-Benzyloxy-4-hvdroxy-phenyl)-3-[3-(tert-butyl-dimethyl-silanyloxy)-3-(4-fluoro- phenyl)-propyπ-1-(4-fluoro-phenyl)-azetidin-2-on 37
3-f3-(tert-Butyl-dimethyl-silanyloxy)-3-(4-fluoro-phenyl)-propyn-4-(2,4-dihydroxy- phenyl)-1 -(4-fluoro-phenyl)-azetidin-2-on 38,
4-(4-Benzyloxy-2-hvdroxy-phenyl)-3-f3-(tert-butyl-dimethyl-silanyloxy)-3-(4-fluoro- phenvD-propyli-i -(4-fluoro-phenyl)-azetidin-2-on 39

Die partielle Hydrierung von Verbindung 36 liefert ein Produktgemisch aus 37:38:39 wie 5.3:1.7:1. Bei einer vollständigen Hydrierung kann man das Diphenol 38 quantitativ erhalten.
13.1 g Lacton 36 werden in 200 ml Ethylacetat gelöst und mit 2 g Pd/C(10%/Pd) 3 Stunden bei 5 bar Wasserstoff hydriert. Das Produktgemisch wird mit
Flashchromatographie (n-Heptan/Ethylacetat 3:1 bis 1 :2) aufgetrennt. Zuerst eluiert Produkt 39, dann 37 und als polarste Verbindung das Produkt 38. Man erhält 5.3 g 37, 1.7 g 38 und 1.0 g 39 alle als amorphe Feststoffe.
Piperazin-1-kohlensäure-3-benzyloxy-4-[3-f3-(tert-butyl-dimethyl-silanyloxy)-3-(4- fluoro-phenyl)-propyπ-1 -(4-fluoro-phenyl)-4-oxo-azetidin-2-vn-phenyl ester 40

1.0 g (1.6 mmol) Phenol 37 werden mit 1.0 g Di-SU-CO analog der Synthese von Verbindung 15 umgestetzt und man erhält 387 mg Piperazinderivat 40 und 405 mg Ausgangsmaterial 37 (dieses bildet sich aus der Zwischenstufe durch Angriff des Piperazins an der phenolischen Seite der Carbonylgruppe.
Piperazin-1 -kohlensaure {4-f3-[3-(tert-butyl-dimethyl-silanyloxy)-3-(4-fluoro- phenyl)- propyll-1-(4-fluoro-phenyl)-4-oxo-azetidin-2-yll-3-hvdroxy-phenyl)-amid 41
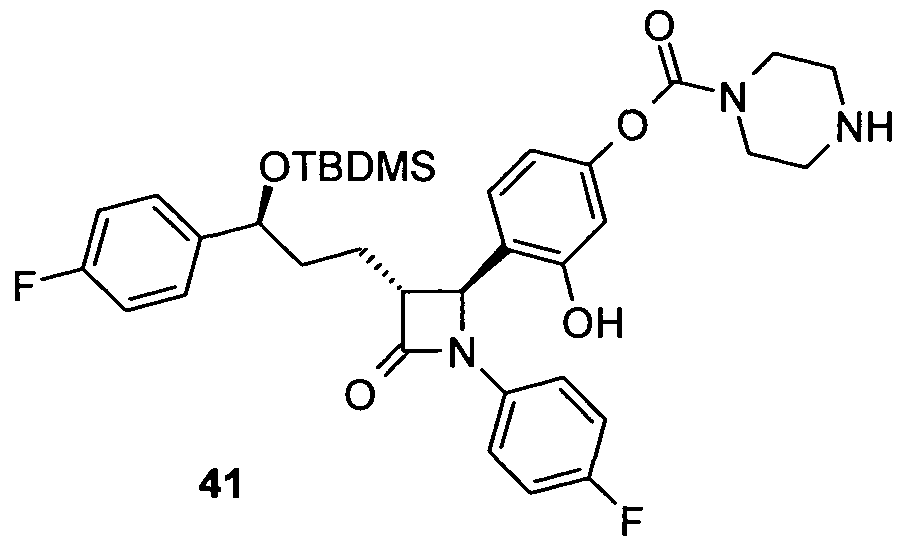
370 mg (0.50 mmol) Verbindung 40 werden analog der Hydrierungsvorschrift von Verbindung 14 benzylentschützt und man erhält 310 mg Piperazinderivat 41 als amorphen Feststoff.
Piperazin-1-kohlensäure-4-{1-(4-fluoro-phenyl)-3-f3-(4-fluoro-phenyl)-3-hvdroxy- propyll-4-oxo-azetidin-2-yl)-3-hvdroxy-phenylester 42

310 mg (0.48 mmol) Verbindung 41 werden mit wässriger HCl entschützt (analog Verbindung 16) und man erhält 124 mg Piperazinderivat 42 als amorphen Feststoff mit dem Molekulargewicht 537.57 (C29H29F2N3O5); MS (ESI+): 538.18 (M+H+).
4-(4-(1-(4-Fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-3-hvdroxy-propyll-4-oxo-azetidin-2-yl)- 3- hvdroxy-phenoxycarbonyl)-piperazin-1-schwefelsäureamid ammoniumsalz 43 (Beispiel I)

70 mg (0.13 mmol) 42 werden mit dem Sθ3-Trimethylamin-Komplex analog der Synthese von Verbindung 17 umgesetzt. Man erhält 77 mg Schwefelsäureamid- derivat 43 (Beispiel I) als amorphen Feststoff mit dem Molekulargewicht 617.16 (C29H29F2N3O8S); MS (ESI+): 1235.05 (2M+H+).
4-Methyl-piperazin-1-kohlensäure-4-(1-(4-fluoro-phenyl)-3-f3-(4-fluoro-phenyl)-3- hvdroxy-propyn-4-oxo-azetidin-2-yl)-3-hvdroxy-phenylester, Verbindung 70

Die Synthese von Verbindung 70 wird analog wie die Herstellung von Verbindung 42 durchgeführt. Anstelle des Piperazins wird Methylpiperazin verwendet. Molekulargewicht 551.60 (C30H3IF2N3O5); MS (ESI+): 1103.08 (2M+H+).
4-(4-(1-(4-Fluoro-phenyl)-3-f3-(4-fluoro-phenyl)-3-hvdroxy-propyn-4-oxo-azetidin-2-yl)- 3- hvdroxy-phenoxycarbonyl)-1.1-dimethyl-piperazin-1-ium Jodid. Beispiel J

56 mg (0.10 mmol) Verbindung 70 werden analog der Synthese von Verbindung Bsp. C mit Methyliodid quarternisiert und man erhält 62 mg Ammoniumsalz Beispiel J als farblosen Feststoff mit dem Molekulargewicht 566.24 (C3i H34F2N3O5); MS (ESI): 566.48 (M+).
Schwefelsäure-mono-(3-{3-benzyloxy-4-f3-f3-(tert-butyl-dimethyl-silanyloxy)-3-(4- fluoro- phenyl)-propyll-1-(4-fluoro-phenyl)-4-oxo-azetidin-2-yll-phenoxy)-propyl)ester- ammoniumsalz 44

1.21 g (1.9 mmol) Phenol 37 werden in 30 ml DMF gelöst. Nach Zugabe von 3.0 g (21 mmol) I .S-Propandiol-cyclic-sulfat (Aldrich) und 4.5 g K2CO3 wird 2 Stunden bei Raumtemperatur gerührt. Die Reaktionslösung wird mit 2 N wässriger HCl leicht sauer gestellt und dann mit Ethylacetat extrahiert. Die organische Phase wird noch 2 mal mit NaCI-Lösung gewaschen, dann mit Methylenchlorid/Methanol/conz. Ammoniak (30/5/1) basisch gestellt und eingeengt. Der Rückstand wird mit Flaschchromatographie (Methylenchlorid/Methanol/conz. Ammoniak 100/7/1 dann 30/5/1 dann 30/10/3) gereinigt und man erhält 1.08 g Amoniumsalz 44.
Schwefelsäure-mono-(3-(4-[3-r3-(tert-butyl-dimethyl-silanyloxy)-3-(4-fluoro-phenyl)- propyll-1-(4-fluoro-phenyl)-4-oxo-azetidin-2-vn-3-hvdroxy-phenoxy)-propyl)ester- ammoniumsalz 45

Schwefelsäure-mono-[3-(4-(1-(4-fluoro-phenyl)-3-f3-(4-fluoro-phenyl)-3-hydroxy- propyπ-4- oxo-azetidin-2-yl)-3-hydroxy-phenoxy)-propynester-ammoniumsalz, Beispiel K

1.5 g Rohprodukt 45 werden 18 Stunden in einem Gemisch aus 40 ml THF und 10 ml 2 N wässriger HCl stehen gelassen. Die Lösung wird mit 50 ml
Methylenchlorid/Methanol/conz. Ammoniak (30/10/3) verdünnt (bis die Lösung basisch ist) und eingeengt. Der Rückstand wird in 30/5/1 suspendiert und über wenig Kieselgel filtriert, mit 30/10/3 nachgewaschen, eingeengt und der erhaltene Rückstand mit Flashchromatographie (Methylenchlorid/Methanol/conz. Ammoniak 30/5/1 dann 30/10/3 dann 30/15/5) getrennt. Man erhält 1.03 g Ammoniumsalz Bsp. K mit dem Molekulargewicht 563.14 (C27H27F2NO8S); MS (ESI+): 1127.14 (2M+H+).
(4-lodo-butoxymethyl)-benzol 46

4-r2-Benzyloxy-4-(4-benzyloxy-butoxy)-phenvn-3-[3-(tert-butyl-dimethyl-silanyloxy)-3- (4-f luoro-phenyl)-propyn-1 -(4-f luoro-phenyl)-azetidin-2-on 47

0.5 g (0.79 mmol) Phenol 37 werden mit 700 mg (2.4 mmol) lodid 46 und 650 mg K2CO3 in 15 ml DMF analog der Synthese von Verbindung 44 alkyliert und man erhält 300 mg 47 als amorphen Feststoff.
3-f3-(tert-Butyl-dimethyl-silanyloxy)-3-(4-fluoro-phenyl)-propyll-1-(4-fluoro-phenyl)-4- r2-hvdroxy-4-(4-hvdroxy-butoxy)-phenvn-azetidin-2-on 48

300 mg (0.38 mmol) 47 werden in 3 ml Methanol mit 60 mg Pd/C (10% Pd) bei 5 bar Wasserstoff 18 Stunden hydriert. Nach Flashchromatographie erhält man 155 mg Alkohol 48.
Schwefelsäure-mono-(4-(4-[3-f3-(tert-butyl-dimethyl-silanyloxy)-3-(4-fluoro-phenyl)- propyll-1-(4-fluoro-phenyl)-4-oxo-azetidin-2-yll-3-hydroxy-phenoxy)-butyl)-ester- ammoniumsalz 49

150 mg (0.25 mmol) 48 werden in 2 ml Pyridin gelöst. Nach Zugabe von 170 mg Trimethylaminschwefeltrioxid-Komplex wird 1 Stunde bei Raumtemperatur gerührt. Dann wird mit 10 ml Methanol und 10 ml Toluol verdünnt und eingeengt. Der Rückstand wird mit Flashchromatographie (Methylenchlorid/Methanol/conz. Ammoniak 30/5/1 dann 30/10/3) gesäult und man erhält 150 mg Sulfat 49.
Schwefelsäure-mono-f4-(4-(1-(4-fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-3-hvdroxy- propyll-4- oxo-azetidin-2-yl}-3-hvdroxy-phenoxy)-butyl1-ester-ammoniumsalz, Beispiel

150 mg (0.22 mmol) 49 werden analog der Synthese von Verbindung Beispiel K TBDMS entschützt und man erhält 69 mg Sulfat Beispiel L mit dem Molekulargewicht 577.19 (C28H29F2NO8S); MS (ESI+): 578.26 (M+H+).
4-r2-Benzyloxy-4-(4-bromo-but-2-enyloxy)-phenyll-3-r3-(tert-butyl-dimethyl-silanyloxy)- 3-(4-fluoro-phenyl)-propyll-1-(4-fluoro-phenyl)-azetidin-2-on 50

200 mg (0.32 mmol) Phenol 37 werden in 5 ml DMF mit 600 mg (2.8 mmol) 1,4- Dibrom-2-buten und 600 mg K2CO3 alkyliert und man erhält 220 mg Bromid 50.
4-r2-Benzyloxy-4-(4-imidazol-1-yl-but-2-enyloxy)-phenyll-3-[3-(tert-butyl-dimethyl- silanyloxy)-3-(4-fluoro-phenyl)-propyn-1-(4-fluoro-phenyl)-azetidin-2-on 51

1-(4-Fluoro-phenyl)-3-r3-(4-fluoro-phenyl)-3-hvdroxy-propyn-4-f2-hydroxy-4-(4- imidazol-1- yl-butoxy)-phenyll-azetidin-2-on, Verbindung 71

3-[4-(4-{1-(4-Fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-3-hvdroxy-propyn-4-oxo-azetidin-2- yl)-3-hvdroxy-phenoxy)-butvH-1-methyl-3H-imidazol-1-ium-iodid, Beispiel M

55 mg (0.10 mmol) 71 werden in 2 ml Toluol gelöst. Nach Zugabe von 2 ml Methyliodid last man 3 Stunden bei 800C Ölbadtemperatur am Rückfluß kochen. Nach Absaugen des ausgefallenen Produktes erhält man 46 mg Bsp. M als farblosen Feststoff mit dem Molekulargewicht 562.25 (C32H34F2N3O4); MS (ESI): 562.11 (M+).
4-r4-Benzyloxy-2-(2-trimethylsilanyl-ethoxymethoxy)-phenvn-3-f3-(tert-butyl-dimethyl- silanyloxy)-3-(4-fluoro-phenyl)-propyn-1-(4-fluoro-phenyl)-azetidin-2-on 53

1.0 g (1.59 mmol) Phenol 39 werden mit 0.8 ml SEMCI (Aldrich) und 1.2 g K2CO3 in 5 ml DMF umgesetzt und man erhält 1.3 g SEM-geschütztes Phenol 53.
3-r3-(tert-Butyl-dimethyl-silanyloxy)-3-(4-fluoro-phenyl)-propyll-1-(4-fluoro-phenyl)-4- [4-hvdroxy-2-(2-trimethylsilanyl-ethoxymethoxy)-phenvn-azetidin-2-on 54

1.1 g (1.4 mmol) 53 werden mit Pd/C analog der Synthese von Verbindung 14 hydriert und man erhält 0.9 g Phenol 54.
4-f4-(4-Bromomethyl-benzyloxy)-2-(2-trimethylsilanyl-ethoxymethoxy)-phenyll-3-[3- (tert- butyl-dimethyl-silanyloxy)-3-(4-fluoro-phenyl)-propyll-1-(4-fluoro-phenyl)-azetidin- 2-on 55

4-f4-(4-Bromomethyl-benzyloxy)-2-hvdroxy-phenyll-1-(4-fluoro-phenyl)-3-f3-(4-fluoro- phenyl)-3-hvdroxy-propyll-azetidin-2-on 56

0.6 g (0.7 mmol) 55 werden in 30 ml THF gelöst. Nach Zugabe von 6 ml 2 N wässriger HCl wird 7 Stunden auf 600C erwärmt. Die Reaktionslösung wird mit 100 ml Ethylacetat verdünnt, die wässrige Phase abgetrennt, die organische Phase noch zwei mal mit NaHCO3-Lösung gewaschen, über wenig Kiselgel filtriert und eingeengt. Der Rückstand wird mit Flashchromatographie (n.Heptan/Ethylacetat 1 :1) getrennt und man erhält 340 mg entschütztes Bromid 56.
1-r4-(4-{1-(4-Fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-3-hvdroxy-propyll-4-oxo-azetidin-2- yl)-3-hvdroxy-phenoxymethyl)-benzvn-4-aza-1-azonia-bicvclor2.2.2loctan-bromid, Beispiel N

122 mg (0.20 mmol) 56 werden in 5 ml Toluol gelöst und mit 150 mg DABCO 2 Stunden bei 800C gerührt. Das ausgefallene Produkt wird abgesaugt, mit Toluol
gewaschen und man erhält 138 mg Trialkylammoniumalkylsalz Bsp. N mit dem Molekulargewicht 640.30 (C38H40F2N3O4); MS (ESI): 640.38 (M+).
Schema 4

59

8 g (33 mmol) Aldehyd 57 und 10 g (Rohprodukt) Anilin 58 werden analog der Synthese von Imin 10 umgesetzt und man erhält 6.5 g kristallines Produkt 59.
(4-[1-(2-Benzyloxy-4-methoxy-phenyl)-5-(tert-butyl-dimethyl-silanyloxy)-5-(4-fluoro- phenyl)-2-(2-oxo-4-phenyl-oxazolidine-3-carbonyl)-pentylaminol-benzyl}- carbaminsäure-benzylester 60

2.5 g (7.0 mmol) Oxazolidinon 11 und 3.0 g (6.2 mmol) Imin 59 werden analog der Synthese von Verbindung 12 umgesetzt und man erhält 1.78 g Produkt 60 und 1.5 g zurückgewonnenes Edukt 11.
(4-{2-(2-Benzyloxy-4-methoxy-phenyl)-3-f3-(tert-butyl-dimethyl-silanyloxy)-3-(4-fluoro- phenyl)-propyn-4-oxo-azetidin-1-yl)-benzyl)-carbaminsäure-benzylester 61

1.78 g (1.9 mmol) 60 werden analog der Synthese von beta-Lactam 13 cyclisiert und man erhält 880 mg Produkt 61.
1-(4-Aminomethyl-phenyl)-3-f3-(4-fluoro-phenyl)-3-hvdroxy-propyll-4-(2-hvdroxy-4- methoxy- phenyl)-azetidin-2-on 62

860 mg (1.1 mmol) 61 werden analog der Synthese von Bsp. O benzyl und silylentschützt und man erhält 300 mg beta-Lactam 62 als amorphen Feststoff.
11-(2, 3,4,5, 6-Pentahvdroxy-hexanoylamino)-undecansäure-4-[3-r3-(4-fluoro-phenyl)-3- hvdroxy-propyll-2-(2-hvdroxy-4-methoxy-phenyl)-4-oxo-azetidin-1-vn-benzylamid, Beispiel O

Bsp. O
60 mg (0.11 mmol) Benzylamin 62 werden anoalg Bsp. G mit 200 mg Säure 63, 150 mg EDC und 75 mg HOBt in 5 ml DMF (16 Stunden bei Raumtemperatur) umgestetzt und man erhält 50 mg acyliertes Bsp O. Dieses Zwischenprodukt wird mit MeOH/NaOMe entschützt und man isoliert 29 mg Zuckerderivat Bsp. O als amorphen Feststoff mit dem Molekulargewicht 811.95 (C43H58FN3O11); MS (ESI+): 812.42 (M+H+).
(4-f3-f3-(4-Fluoro-phenyl)-3-hvdroxy-propyn-2-(2-hvdroxy-4-methoxy-phenyl)-4-oxo- azetidin-1-vπ-benzyl)-carbaminsäure-2,5-dioxo-pyrrolidin-1-yl-ester 64

100 mg (0.22 mmol) 62 werden in 5 ml Acetonitil gelöst und mit 100 mg Di-Su-CO 1 Stunde bei Raumtemperatur gerührt. Nach aufkonzentrieren der Reaktionslösung wird der Rückstand mit Flashchromatographie (n-Heptan/Ethylacetat 1 :2 dann 0:1) gereinigt und man erhält 60 mg 64.
1-r2-(4-{4-[3-r3-(4-Fluoro-phenyl)-3-hvdroxy-propyn-2-(2-hvdroxy-4-methoxy-phenyl)-4- OXO- azetidin-1 -yll-benzylcarbamoyl)-piperazin-1 -yl)-2-oxo-ethyll-4-aza-1 -azonia- bicyclo[2.2.21octane-chlorid Beispiel P

60 mg (0.10 mmol) 64 werden in 3 ml Acetonitril gelöst. Nach Zugabe von 40 mg Piperazinderivat 65 und 0.3 ml 1 N wässrige Natronlauge rührt man 4 Stunden bei Raumtemperatur. Die Reaktionslösung wird auf 1 ml eingengt, mit 1 ml DMF verdünnt und über präparative HPLC gereinigt. Man erhält 23 mg Ammoniumsalz Bsp. P mit dem Molekulargewicht 715.36 (C39H48FN6O6); MS (ESI):715.34 (M+).
Die erfindungsgemäßen Verbindungen der Formel I wurden mit der nachfolgend beschriebenen Methode auf ihre Wirkung geprüft:
Beeinflussung der Cholesterolabsorption + 3H- Taurocholsäureausscheidung anhand der fäkalen Ausscheidung an der Maus, Ratte oder Hamster
NMRI- Mäuse, Wistar-Ratten, oder Golden Syrian Hamster (in Gruppen von n=4-6) werden unter Standarddiät (Altromin, Lage (Lippe)) in Stoffwechselkäfigen gehalten. Am Nachmittag vor Gabe der radioaktiven Tracer (14C-Cholesterol) werden die Tiere nüchtern gesetzt und auf Gitterroste adaptiert.
Zusätzlich werden die Tiere werden 24 Stunden vor der peroralen Applikation der Testmahlzeit (14C-Cholesterol in Intralipid® 20, Pharmacia-Upjohn) mit 3H-TCA (Taurocholic acid) s.c. gelabelt (z.B. 1 μCi/Maus bis 5 μCi/Ratte)
Cholesterolabsorptionstest: 0,25 ml/Maus Intralipid ® 20 (Pharmacia- Upjohn) ((Spikung mit 0,25 μCi 14C-Cholesterol in 0,1 mg Cholesterol) werden peroral mit der Schlundsonde verabreicht.
Testsubstanzen werden getrennt in 0,5 %/ (Methylcellulose (Sigma)/5% Solutol (BASF, Ludwigshafen ) oder geeignetem Vehikel angesetzt. Das Applikationsvolumen der Testsubstanz beträgt 0,5 ml /Maus. Die Testsubstanz wird unmittelbar vor der Testmahlzeit (Intralipid mit 14C-Cholesterol-label) (Cholesterolabsorptionstest) oral appliziert.
Der Kot wird über 24 h gesammelt: die fäkale Elimination von 14C-Cholesterol und 3H Taurocholsäure (TCA) nach 24 Std. wird bestimmt.
Die Lebern werden entnommen, homogenisiert und Aliquots im Oximaten (Model 307, P Paacckkaarrdd)) vveerbrannt zur Bestimmung der aufgenommenn/resorbierten Menge an 14C- Cholesterol.
Auswertung: Kotproben:
Es wird das Gesamtgewicht bestimmt, mit Wasser auf definiertes Volumen aufgefüllt, dann homogenisiert, Aliquot eingetrocknet und im Oximat (Model 307, Packard zur Verbrennung von radioaktiv gelabelten Proben) verbrannt: Die Menge von radioaktiv 3H- H2O und 14C- CO2 wird hochgerechnet auf die ausgeschiedene Menge an 3H- Taurocholsäure bzw. 14C-Cholesterol (Dual-Isotopen-Technik ). Die ED2Oo-Werte werden als Dosis aus einer Dosiswirkungskurve interpoliert als diejenige Dosen, die die Auscheidung an TCA bzw. Cholesterol verdoppeln, bezogen auf eine zeitgleich behandelte Kontrollgruppe.
Leberproben:
Die aufgenommene Menge von 14C-Cholesterols in die Leber wird in Abhängigkeit von der applizierten Dosis der Testsubstanz im Verhältnis zu einer Kontrollgruppe (Vehikel-behandelt) ausgewertet. Die ED50 Werte werden interpoliert aus einer Dosiswirkungskurve als diejenige Dosis, die die Aufnahme von 14C- Cholesterol in die Leber halbiert (50%), bezogen auf eine Kontrollgruppe.
Die folgenden ED50 -Werte (Leberwerte; Maus) belegen die Aktivität der erfindungsgemäßen Verbindungen der Formel I
Beispiel Nr. EDsn (Leber) [mq/Mausl
0 < 0.01 M < 0.1 A 0.01 P 0.3 B < 0.1
1 0.01 H 0.01 F 0.3 E < 0.01 C 0.1
K < 0.01
N 0.01
Aus der Tabelle ist abzulesen, daß die Verbindungen der Formel I sehr gut die Aufnahme von Cholesterol aus dem Magendarmtrakt hemmen.
Hamstermodell:
Syrische Hamster (in Gruppen von n=5-9) erhalten eine mit 0,1 % cholesterinangereicherter Standarddiät (ssniff, Soest Germany).
Die Testsubstanzen werden in 0,5 %/ (Methylcellulose (Sigma)/5% Solutol (BASF, Ludwigshafen) oder einem anderen geeignetem Vehikel angesetzt und mindestens 3 Dosierungen an 12 aufeinader folgenden Tagen einmal täglich mit der Schlundsonde appliziert. Am Tag 12 werden die Tiere in tiefer Narkose aus der Aorta entblutet. Im Serum werden Gesamt-Cholesterin, LDL- Cholesterin, HDL-Cholesterin und Triglyceride mit Standard Kits von Roche nach den Richtlinen der German Society for Clinical
Chemistry analysiert.
Die ED50 Werte für LDL Cholesterincholesterinsenkung gegenüber placebobehandelten Kontrolltieren wurden mit einem Standard logistischen Modell für die Dosis-Wirkungs-Kurve berechnet.
Daten aus Hamsterversuchen:
Die folgenden ED50 -Werte (Serum LDL Cholesterinwerte; Hamster, [mg/kg]) belegen die Aktivität der erfindungsgemäßen Verbindungen der Formel I:
Beispiel Nr. ED50
A 0.2
I <0.1
E 0.1
Leberexposition:
Die Leberexposition der erfindungsgemäßen Verbindungen gegenüber den entsprechenden Verbindungen ohne Hydroxyfunktion in 2"-Stellung wurde in vivo an der männlichen Wistar-Ratte untersucht. An der mit Ketamin/Midazolam (Ketamin 80 mg/kg i.p. + Midazolam 5 mg/kg i.p.) betäubten Ratte wird nach Laparotomie in der Linea alba das Präparat intraduodenal appliziert. Hierbei wird versucht, einen unmittelbaren Reflux des Präparates in den Magen zu verhindern. Die Tiere bleiben während des gesamten Versuches in Narkose. Am Versuchsende, nach 2 h, wird für die Substanzbestimmung die Leber herauspräpariert. Die Bestimmung der Substanzspiegel in den Leberhomogenaten erfolgt mittels LC-MS/MS. Hierzu werden zunächst die Proteine durch Zugabe von Acetoniril in Anwesenheit eines internen Standards ausgefällt. Ein Teil des Überstands wird mit einem geeignetem Puffer versetzt, und ein Aliquot der Mischung wird injiziert. Die Auswertung der Messung erfolgt über die Peakflächen von Analyt und internem Standard.
Ergebnisse:
Cmax-Werte in der Leber (L) nach oraler Gabe von 10 mg/kg Körpergewicht
"H-Verbindungen" "2"-OH-Verbindungen"
Beispiel K


L = 24.3 μg/mL L = 0.30 μg/mL
Beispiel N


L = 0.20 μg/mL L = 0.08 μg/mL
Aus diesen Daten ist abzulesen, dass die erfindungsgemäßen Verbindungen deutlich geringere Leberspiegel aufweisen und so die Belastung der Leber reduzieren.
Claims
1. Verbindungen der Formel I1

worin bedeuten
R1 , R2, R3, R4, R5, R6 unabhängig voneinander (Ci-C30)-Alkylen-(LAG)n, wobei n = 1 - 5 sein kann und wobei ein oder mehrere C-Atome des
Alkylenrests durch -S(O)n-, mit n = 0 - 2, -O-, -(C=O)-, -(C=S)-, -CH=CH-, -C≡C-, -N((Ci-C6)-Alkyl)-, -N(Phenyl)-, -N((CrC6)-Alkyl-Phenyl)-, -N(CO- (CH2)i-io-COOH)-, -N(CO-(CrC8)-Alkyl)-, -N(CO-(C3-C8)-Cycloalkyl), N(CO-(CH2)o-io-Aryl), -N(CO-(CH2)o-io-Heteroaryl), -NH- oder durch bis zu dreifach mit R7 substituierte Aryl- oder Heteroarylreste oder durch bis zu vierfach mit R7 substituierte (C3-Ci 0)-Cycloalkyl- oder Heterocycloalkylreste ersetzt sein können;
H, F, Cl, Br, I, CF3, NO2, N3, CN, COOH, COO(d-C6)Alkyl, CONH2, CONH(C1-C6)Alkyl, CON[(Ci-C6)Alkyl]2, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2- C6)-Alkinyl, O-(Ci-C6)-Alkyl, wobei in den Alkylresten ein, mehrere, oder alle Wasserstoff(e) durch Fluor ersetzt sein können; C(=NH)(NH2), PO3H2, SO3H, SO2-NH2, SO2NH(CrC6)-Alkyl, SO2N[(Cr C6)-Alkyl]2 , S-(Ci-C6)-Alkyl, S-(CH2)n-Phenyl, SO-(C1-C6)-Alkyl, SO- (CH2)n-Phenyl, SO2-(C1-C6)-Alkyl, SO2-(CH2)n-Phenyl, wobei n = O - 6 sein kann und der Phenylrest bis zu zweifach mit F, Cl, Br, OH, CF3, NO2, CN, OCF3, O-(Ci-C6)-Alkyl, (CrC6)-Alkyl, NH2 substituiert sein kann; NH2, NH-(Ci-C6)-Alkyl, N((CrC6)-Alkyl)2, NH(Ci-C7)-Acyl, Phenyl, O- (CH2)n-Phenyl, wobei n = O - 6 sein kann, wobei der Phenylring ein bis 3- fach substituiert sein kann mit F, Cl, Br, I, OH, CF3, NO2, CN, OCF3, O-
(Ci-C6)-Alkyl, (CrC6)-Alkyl, NH2, NH(CrC6)-Alkyl, N((CrC6)-Alkyl)2, SO2- CH3, COOH, COO-(Ci-C6)-Alkyl, CONH2;
R7 F, Cl, Br, I, CF3, NO2, N3, CN, COOH, COOCd-CeJAlkyl, CONH2, CONH^-CeOAlkyl, CONKd-CeJAlkylk, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2-
C6)-Alkinyl, O-(Ci-C6)-Alkyl, wobei in den Alkylresten ein, mehrere, oder alle Wasserstoff(e) durch Fluor ersetzt sein können; C(=NH)(NH2), PO3H2, SO3H, SO2-NH2, SO2NH(CrC6)-Alkyl, SO2Nt(C1- C6)-Alkyl]2 , S-(Ci-C6)-Alkyl, S-(CH2)n-Phenyl, SO-(d-C6)-Alkyl, SO- (CH2)n-Phenyl, SO2-(d-C6)-Alkyl, SO2-(CH2)n-Phenyl, wobei n = O - 6 sein kann und der Phenylrest bis zu zweifach mit F, Cl, Br, OH, CF3, NO2, CN, OCF3, O-(CrC6)-Alkyl, (Ci-C6)-Alkyl, NH2 substituiert sein kann; NH2, NH-(C1-C6)-Alkyl, N((Ci-C6)-Alkyl)2, NH(Ci-C7)-Acyl, Aryl, O-(CH2)n- Aryl, wobei n = O - 6 sein kann, wobei der Arylring ein bis 3-fach substituiert sein kann mit F, Cl, Br, I, OH, CF3, NO2, CN, OCF3, 0-(Ci-C6)-
Alkyl, (Ci-C6)-Alkyl, NH2, NH(C1-C6)-Alkyl, N((C1-C6)-Alkyl)2, SO2-CH3, COOH, COO-(Ci-C6)-Alkyl, CONH2;
LAG C4-Cio-cycloaliphatischer mit 2 bis 9 Hydroxyfunktionen substituierter Rest oder Cz-do-aliphatischer mit 2 bis 10 Hydroxyfunktionen substituierter Rest, wobei jeweils eine oder mehrere Hydroxyfunktionen durch einen -NHR8-Rest ersetzt sein können;
Aminosäurerest, Oligopeptidrest bestehend aus 2 bis 9 Aminosäuren; acyclischer, mono- oder bi-cyclischer Trialkylammonium-Rest, acyclischer, mono- oder bicyclischer Thalkylammoniumalkyl-Rest, wobei bis zu drei Kohlenstoffatome durch N, O oder S(O), mit n = 0-2, ersetzt sein können; N-alkylierte Heteroaromaten, wie z. B. Imidazolium oder Pyridinium;
-O-(SO2)-OH; -(CH2)O-IO-SO3H; -(CH2)o-io-P(0)(OH)2, -(CH2)o-io-0- P(O)(OH)2, -(CHz)o.io-C(=NH)(NH2); -(CH2)0-io-C(=NH)(NHOH); -NR8- C(=NR9)(NR10R11); wobei n = 1 - 5 ist und R8, R9, R10 und R11 unabhängig voneinander H, (Ci-C6)-Alkyl, Phenyl, (CrC6)-Alkyl-Phenyl,
(C3-C8)-Cycloalkyl), -C(O)-(Ci-C6)-Alkyl, -C(O)-(C3-C8)-Cycloalkyl sein können;
wobei immer mindestens einer der Reste R1 bis R6 die Bedeutung (Ci-C3o)-Alkylen-(LAG)n, wobei n = 1 - 5 sein kann und wobei ein oder mehrere C-
Atome des Alkylenrests durch -S(O)n-, mit n = O - 2, -O-, -(C=O)-, -(C=S)-, -CH=CH-, - C≡C-, -N((Ci-C6)-Alkyl)-, -N(Phenyl)-, -N((C1-C6)-Alkyl-Phenyl)-, -N(CO-(CH2)1-10- COOH)-, -N(CO-(Ci-C8)-Alkyl)-, -N(CO-(C3-C8)-Cycloalkyl), N(CO-(CH2)o-io-Aryl), - N(CO-(CH2)0-io-Heteroaryl), -NH- oder durch bis zu dreifach mit R7 substituierte Aryl- oder Heteroarylreste oder durch bis zu vierfach mit R7 substituierte (C3-Ci0)- Cycloalkyl- oder Heterocycloalkylreste ersetzt sein können; besitzen muß,
sowie deren pharmazeutisch verträglichen Salze.
2. Verbindungen der Formel I, gemäß Anspruch 1 , dadurch gekennzeichnet, dass darin bedeuten
R1 , R2, R3, R4, R5, R6 unabhängig voneinander (Ci-C20)-Alkylen-(LAG), wobei ein oder mehrere C-Atome des Alkylenrests durch -O-, -(C=O)-, - N((C1-C6)-Alkyl)-, -N(CO-(CH2)LIO-COOH)- oder -NH- oder durch bis zu dreifach mit R7 substituierte Aryl- oder Heteroarylreste oder durch bis zu vierfach mit R7 substituierte (C3-Ci 0)-Cycloalkyl- oder Heterocycloalkylreste ersetzt sein können;
H, F, Cl, Br, I, CF3, NO2, N3, CN, COOH, COO(Ci-C6)Alkyl, CONH2,
CONH(Ci-C6)Alkyl, CON[(d-C6)Alkyl]2, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2- C6)-Alkinyl, O-(CrC6)-Alkyl, wobei in den Alkylresten ein, mehrere, oder alle Wasserstoff(e) durch Fluor ersetzt sein können; C(=NH)(NH2), PO3H2, SO3H, SO2-NH2, SO2NH(d-C6)-Alkyl, SO2Nf(C1- C6)-Alkyl]2 , S-(d-C6)-Alkyl, S-(CH2)n-Phenyl, SO-(Ci-C6)-Alkyl, SO- (CH2)n-Phenyl, SO2-(d-C6)-Alkyl, SO2-(CH2)n-Phenyl, wobei n = O - 6 sein kann und der Phenylrest bis zu zweifach mit F, Cl, Br, OH, CF3, NO2,
CN, OCF3, O-(C1-C6)-Alkyl, (C1-C6)-Alkyl, NH2 substituiert sein kann; NH2, NH-(C1-C6)-Alkyl, N((d-C6)-Alkyl)2, NH(CrC7)-Acyl, Phenyl, O- (CH2)n-Phenyl, wobei n = O - 6 sein kann, wobei der Phenylring ein bis 3- fach substituiert sein kann mit F, Cl, Br, I, OH, CF3, NO2, CN, OCF3, O- (Ci-C6)-Alkyl, (CrC6)-Alkyl, NH2, NH(C1-Ce)-AIKyI1 N((Ci-C6)-AIKyl)2, SO2-
CH3, COOH, COO-(CrC6)-Alkyl, CONH2;
R7 F, Cl, Br, I1 CF3, NO2, N3, CN, COOH, COO(Ci-C6)Alkyl, CONH2,
CONH(Ci-C6)AIKyI1 CON[(Ci-C6)AIKyl]2, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2- C6)-Alkinyl, O-(CrC6)-Alkyl, wobei in den Alkylresten ein, mehrere, oder alle Wasserstoff(e) durch Fluor ersetzt sein können; C(=NH)(NH2), PO3H2, SO3H, SO2-NH2, SO2NH(Ci-Ce)-AIKyI1 SO2Nf(C1- C6J-AIKyI]2 , S-(Ci-Ce)-AIKyI1 S-(CH2)n-Phenyl, SO-(d-C6)-Alkyl, SO- (CH2)n-Phenyl, SO2-(d-C6)-Alkyl, SO2-(CH2)n-Phenyl, wobei n = O - 6 sein kann und der Phenylrest bis zu zweifach mit F, Cl, Br, OH, CF3, NO2,
CN, OCF3, O-(Ci-C6)-Alkyl, (CrC6)-Alkyl, NH2 substituiert sein kann; NH2, NH-(Ci-Ce)-AIKyI1 N((Ci-C6)-AIKyl)2, NH(d-C7)-Acyl, Aryl, O-(CH2)n- Aryl, wobei n = O - 6 sein kann, wobei der Arylring ein bis 3-fach substituiert sein kann mit F, Cl, Br, I, OH, CF3, NO2, CN, OCF3, 0-(C1-C6)- AIKyI1 (C1-Ce)-AIKyI, NH2, NH(CrCe)-AIKyI1 N((d-C6)-Alkyl)2, SO2-CH3,
COOH, COO-(CrCe)-AIKyI1 CONH2;
LAG C4-Cio-cycloaliphatischer mit 2 bis 9 Hydroxyfunktionen substituierter
Rest oder C2-do-aliphatischer mit 2 bis 10 Hydroxyfunktionen substituierter Rest, wobei jeweils eine oder mehrere Hydroxyfunktionen durch einen -NHR8-Rest ersetzt sein können; Aminosäurerest, Oligopeptidrest bestehend aus 2 bis 9 Aminosäuren; acyclischer, mono- oder bi-cyclischer Trialkylammonium-Rest, acyclischer, mono- oder bicyclischer Trialkylammoniumalkyl-Rest, wobei bis zu drei Kohlenstoffatome durch N, O oder S(O), mit n = 0-2, ersetzt sein können; N-alkylierte Heteroaromaten, wie z. B. Imidazolium oder Pyridinium;
-O-(SO2)-OH; -(CH2)O-I0-SO3H; -(CH2)o-io-P(0)(OH)2, -(CH2)0-io-O- P(O)(OH)2, -(CH2)o-io-C(=NH)(NH2); -(CH2)0-io-C(=NH)(NHOH); -NR8- C(=NR9)(NR10R11 ); wobei n = 1 - 5 ist und R8, R9, R10 und R11 unabhängig voneinander H, (Ci-C6)-Alkyl, Phenyl, (Ci-CβJ-Alkyl-Phenyl, (C3-C8)-Cycloalkyl), -C(O)-(CrC6)-Alkyl, -C(O)-(C3-C8)-Cycloalkyl sein können;
wobei immer mindestens einer der Reste R1 bis R6 die Bedeutung (Ci-C20)-Alkylen-(LAG), wobei ein oder mehrere C-Atome des Alkylenrests durch -O-, -(C=O)-, -N((Ci-C6)-Alkyl)-, -N(CO-(CH2)I-Io-COOH)- oder -NH- oder durch bis zu dreifach mit R7 substituierte Aryl- oder Heteroarylreste oder durch bis zu vierfach mit R7 substituierte (C3-Ci0)-Cycloalkyl- oder Heterocycloalkylreste ersetzt sein können; besitzen muß,
sowie deren physiologisch verträglichen Salze.
3. Verbindungen der Formel I, gemäß Anspruch 1 oder 2, dadurch gekennzeichnet, dass darin bedeuten
R1 , R3 unabhängig voneinander H oder (Ci-Ci2)-Alkylen-(LAG) hat, wobei ein oder mehrere C-Atome des Alkylenrests durch -O-, -(C=O)-, -N(CH3)-, oder -NH- oder durch bis zu dreifach mit R7 substituierte Aryl- oder Heteroarylreste oder durch bis zu vierfach mit R7 substituierte (C3-C10)- Cycloalkyl- oder Heterocycloalkylreste ersetzt sein können, wobei immer einer der Reste R1 und R3 immer die Bedeutung H und der andere die
Bedeutung (Ci-Ci2)-Alkylen-(LAG) besitzt. R2, R4, R5, R6 H;
R7 F, Cl, Br, I1 CF3, NO2, N3, CN, COOH, COO(Ci-C6)Alkyl, CONH2,
CONH(CrC6)Alkyl, CON[(d-C6)Alkyl]2, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2- C6)-Alkinyl, O-(Ci-C6)-Alkyl, wobei in den Alkylresten ein, mehrere, oder alle Wasserstoff(e) durch Fluor ersetzt sein können; C(=NH)(NH2), PO3H2, SO3H, SO2-NH2, SO2NH(C1-C6)-Alkyl, SO2N[(Cr C6)-Alkyl]2 , S-(C1-C6)-Alkyl, S-(CH2)n-Phenyl, SO-(Ci-C6)-Alkyl, SO- (CH2)n-Phenyl, SO2-(C i-C6)-Alkyl, SO2-(CH2)n-Phenyl, wobei n = O - 6 sein kann und der Phenylrest bis zu zweifach mit F, Cl, Br, OH, CF3, NO2,
CN, OCF3, O-(Ci-C6)-Alkyl, (Ci-C6)-Alkyl, NH2 substituiert sein kann; NH2, NH-(Ci-C6)-Alkyl, N((Ci-C6)-Alkyl)2, NH(C1-Cy)-ACyI, Aryl, O-(CH2)n- Aryl, wobei n = O - 6 sein kann, wobei der Arylring ein bis 3-fach substituiert sein kann mit F, Cl, Br, I1 OH, CF3, NO2, CN, OCF3, 0-(C1-C6)- Alkyl, (CrC6)-Alkyl, NH2, NH(C1-C6)-Alkyl, N((C1-C6)-Alkyl)2, SO2-CH3,
COOH, COO-(Ci-C6)-Alkyl, CONH2;
LAG C4-C10-cycloaliphatischer mit 2 bis 9 Hydroxyfunktionen substituierter
Rest oder C2-Cio-aliphatischer mit 2 bis 10 Hydroxyfunktionen substituierter Rest, wobei jeweils eine oder mehrere Hydroxyfunktionen durch einen -NHR8-Rest ersetzt sein können;
Aminosäurerest, Oligopeptidrest bestehend aus 2 bis 9 Aminosäuren; acyclischer, mono- oder bi-cyclischer Trialkylammonium-Rest, acyclischer, mono- oder bicyclischer Trialkylammoniumalkyl-Rest, wobei bis zu drei Kohlenstoffatome durch N, O oder S(O), mit n = 0-2, ersetzt sein können; N-alkylierte Heteroaromaten, wie z. B. Imidazolium oder
Pyridinium;
-O-(SO2)-OH; -(CH2)O-10-SO3H; -(CH2)0-10-P(O)(OH)2, -(CH2)0-10-O-
P(O)(OH)2, -(CH2)0-10-C(=NH)(NH2); -(CH2)o-io-C(=NH)(NHOH); -NR8- C(=NR9)(NR10R11); wobei n = 1 - 5 ist und R8, R9, RIO und R11 unabhängig voneinander H, (Ci-C6)-Alkyl, Phenyl, (d-C^-Alkyl-Phenyl,
(C3-C8)-Cycloalkyl), -C(O)-(Ci-C6)-Alkyl, -C(O)-(C3-C8)-Cycloalkyl sein können;
sowie deren physiologisch verträglichen Salze.
4. Verbindungen der Formel I, gemäß einem oder mehreren der Ansprüche 1 bis 3, dadurch gekennzeichnet, dass darin bedeuten
LAG -(CH2)o-io-0-(Sθ2)-OH, -(CH2)o-io-S03H oder ein mono- oder bi- cyclischer Trialkylammoniumalkyl-Rest, in dem bis zu drei Kohlenstoffatome durch N, O oder S(O)n, mit n = 0-2, ersetzt sein können;
sowie deren physiologisch verträglichen Salze.
5. Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 4 zur Anwendung als Arzneimittel.
6. Arzneimittel enthaltend eine oder mehrere der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 4.
7. Arzneimittel enthaltend eine oder mehrere der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 4 und mindestens einen weiteren Wirkstoff.
8. Arzneimittel, gemäß Anspruch 7, dadurch gekennzeichnet, dass es als weiteren Wirkstoff eine oder mehrere Antidiabetika, hypoglykämischen Wirkstoffe, HMGCoA-
Reduktase Inhibitoren, Cholesterinresorptionsinhibitoren, PPAR gamma Agonisten, PPAR alfa Agonisten, PPAR alfa/gamma Agonisten, PPAR delta Agonisten, Fibrate, MTP-Inhibitoren, Gallensäureresorptionsinhibitoren, MTP-Inhibitoren, CETP- Inhibitoren, polymere Gallensäureadsorber, LDL-Rezeptorinducer, ACAT-Inhibitoren, Antioxidantien, Lipoprotein-Lipase Inhibitoren, ATP-Citrat-Lyase Inhibitoren, Squalen synthetase inhibitoren, Lipoprotein(a) antagonisten, HM74A Rezeptor Agonisten, Lipase Inhibitoren, Insuline, Sulfonylharnstoffe, Biguanide, Meglitinide, Betazellen wirkende Wirkstoffe, Glycogenphosphorylaseinhibitoren, Glukagon- Rezeptor-Antagonisten, Aktivatoren der Glukokinase, Inhibitoren der Glukoneogenese, Inhibitoren der Fructose-1 ,6-biphosphatase, Modulatoren des Glukosetransporters-4, Inhibitoren der Glutamin-Fructose-6-Phosphat-Amidotransferase, Inhibitoren der Dipeptidylpeptidase-IV, Hemmstoffe der 11-beta-Hydroxysteroid-Dehydrogenase-1 , Inhibitoren der Protein-Tyrosin-Phosphatase-1 B, Modulatoren des natrium-abhängigen Glukosetransporters 1 oder 2, Modulatoren des GPR40, Inhibitoren der hormonsensitiven Lipase, Hemmstoffe der der Acetyl-CoA Carboxylase, Inhibitoren der Phosphoenolpyruvatcarboxykinase, Inhibitoren der Glykogen Synthase Kinase-3 beta, Inhibitoren der Protein Kinase C beta, Endothelin-A-Rezeptor Antagonisten,
Inhibitoren der I kappaB kinase, Modulatoren des Glucocorticoidrezeptors, CART- Agonisten, NPY-Agonisten, MC4-Agonisten, Orexin-Agonisten, H3-Agonisten, TNF- Agonisten, CRF-Agonisten, CRF BP-Antagonisten, Urocortin-Agonisten, ß3-Agonisten, , CB1 -Rezeptor Antagonisten ,MSH (Melanocyt-stimulierendes Hormon)-Agonisten, CCK-Agonisten, Serotonin-Wiederaufnahme-Inhibitoren, gemischte Sertonin- und noradrenerge Verbindungen, 5HT-Agonisten, Bombesin-Agonisten, Galanin- Antagonisten, Wachstumshormone, Wachstumshormon freisetzende Verbindungen, TRH-Agonisten, entkoppelnde Protein 2- oder 3-Modulatoren, Leptinagonisten, DA- Agonisten (Bromocriptin, Doprexin), Lipase/Amylase-Inhibitoren, PPAR-Modulatoren, RXR-Modulatoren oder TR-ß-Agonisten oder Amphetamine enthält.
9. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 4 zur Herstellung eines Medikamentes zur Blutzuckersenkung.
10. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 4 zur Herstellung eines Medikamentes zur Behandlung des Typ Il Diabetes.
11. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 4 zur Herstellung eines Medikamentes zur Behandlung von Lipid- und Kohlenhydratstoffwechselstörungen.
12. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 4 zur Herstellung eines Medikaments zur Behandlung arteriosklerotischer Erscheinungen.
13. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 4 zur Herstellung eines Medikaments zur Behandlung von Insulin Resistenz.
14. Verfahren zur Herstellung eines Arzneimittels enthaltend eine oder mehrere der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 4, dadurch gekennzeichnet, dass der Wirkstoff mit einem pharmazeutisch geeigneten Träger vermischt wird und diese Mischung in eine für die Verabreichung geeignete Form gebracht wird.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008541615A JP2009516714A (ja) | 2005-11-23 | 2006-11-13 | 高脂血症を治療するためのヒドロキシ置換ジフェニルアゼチジノン |
| EP06829012A EP1954673A1 (de) | 2005-11-23 | 2006-11-13 | Hydroxy-substituierte diphenylazetidinone zur behandlung von hyperlipidämie |
| US12/122,991 US20080274947A1 (en) | 2005-11-23 | 2008-05-19 | Hydroxy-Substituted Diphenylazetidinones for the Treatment of Hyperlipidemia |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005055726A DE102005055726A1 (de) | 2005-11-23 | 2005-11-23 | Hydroxy-substituierte Diphenylazetidinone, Verfahren zu deren Herstellung, diese Verbindungen enthaltende Arzneimittel und deren Verwendung |
| DE102005055726.0 | 2005-11-23 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/122,991 Continuation US20080274947A1 (en) | 2005-11-23 | 2008-05-19 | Hydroxy-Substituted Diphenylazetidinones for the Treatment of Hyperlipidemia |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007059871A1 true WO2007059871A1 (de) | 2007-05-31 |
Family
ID=37507317
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/010840 WO2007059871A1 (de) | 2005-11-23 | 2006-11-13 | Hydroxy-substituierte diphenylazetidinone zur behandlung von hyperlipidämie |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20080274947A1 (de) |
| EP (1) | EP1954673A1 (de) |
| JP (1) | JP2009516714A (de) |
| DE (1) | DE102005055726A1 (de) |
| WO (1) | WO2007059871A1 (de) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2009140932A2 (en) * | 2008-05-21 | 2009-11-26 | Zentiva, K.S. | Method of producing (3r,4s)-l-(4-fluorophenyl)-3-[(3s)-3-(4-fluorophenyl)- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone |
| US7842684B2 (en) | 2006-04-27 | 2010-11-30 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| US7863265B2 (en) | 2005-06-20 | 2011-01-04 | Astrazeneca Ab | 2-azetidinone derivatives and their use as cholesterol absorption inhibitors for the treatment of hyperlipidaemia |
| US7871998B2 (en) | 2003-12-23 | 2011-01-18 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7893048B2 (en) | 2005-06-22 | 2011-02-22 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7906502B2 (en) | 2005-06-22 | 2011-03-15 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2012004382A1 (en) * | 2010-07-09 | 2012-01-12 | Moehs Iberica S.L. | New method for preparing ezetimibe |
| WO2012030165A2 (ko) | 2010-08-31 | 2012-03-08 | 서울대학교산학협력단 | P P A R δ 활성물질의 태자재프로그래밍용도 |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| EP2567959A1 (de) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridin-4-Carbonsäureamid-Derivate als Kinaseinhibitoren |
| WO2014093189A1 (en) | 2012-12-10 | 2014-06-19 | Merck Sharp & Dohme Corp. | Methods of treating diabetes by administering a glucagon receptor antagonist in combination with a cholesterol absorption inhibitor |
| WO2017050990A1 (fr) * | 2015-09-25 | 2017-03-30 | Universite De Nantes | 1,4-di-(4-methylthiophenyl)-3-phtaloylazetidine-2-one et ses derives |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002050027A1 (de) * | 2000-12-21 | 2002-06-27 | Aventis Pharma Deutschland Gmbh | Neue diphenzylazetidinone, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung zur behandlung von lipidstoffwechselstörungen |
| US7671047B2 (en) * | 2002-06-19 | 2010-03-02 | Sanofi-Aventis Deutschland Gmbh | Cationically substituted diphenylazetidinones, process for their preparation, medicaments comprising these compounds, and their use |
| CA2545058A1 (en) * | 2003-11-10 | 2005-05-26 | Microbia, Inc. | 4-biarylyl-1-phenylazetidin-2-ones |
| TW200806623A (en) * | 2005-10-05 | 2008-02-01 | Merck & Co Inc | Anti-hypercholesterolemic compounds |
| KR20090091120A (ko) * | 2006-11-02 | 2009-08-26 | 사노피-아벤티스 도이칠란트 게엠베하 | 향상된 약리학적 특성을 갖는 피페라진-1-설폰산으로 치환된 신규한 디페닐아제티디논 |
| DE102007063671A1 (de) * | 2007-11-13 | 2009-06-25 | Sanofi-Aventis Deutschland Gmbh | Neue kristalline Diphenylazetidinonhydrate, diese Verbindungen enthaltende Arzneimittel und deren Verwendung |
| US8530460B2 (en) | 2011-12-19 | 2013-09-10 | Boehringer Ingelheim International Gmbh | Azetidine derivatives |
| US8530461B2 (en) * | 2011-12-29 | 2013-09-10 | Boehringer Ingelheim International Gmbh | Azetidine derivatives |
| US8623860B2 (en) | 2011-12-30 | 2014-01-07 | Boehringer Ingelheim International Gmbh | Azetidine derivatives, pharmaceutical compositions and uses thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005047248A1 (en) * | 2003-11-10 | 2005-05-26 | Microbia, Inc. | 4-biarylyl-1-phenylazetidin-2-ones |
| WO2005069900A2 (en) * | 2004-01-16 | 2005-08-04 | Merck & Co., Inc. | Npc1l1 (npc3) and methods of identifying ligands thereof |
| WO2006086562A2 (en) * | 2005-02-09 | 2006-08-17 | Microbia, Inc. | Phenylazetidinone derivatives |
| WO2006116499A1 (en) * | 2005-04-26 | 2006-11-02 | Microbia, Inc. | 4-biarylyl-1-phenylazetidin-2-one glucuronide derivatives for hypercholesterolemia |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BRPI0611415A2 (pt) * | 2005-05-25 | 2010-09-08 | Microbia Inc | ácidos fosfÈnicos de 4-(bifenilil)azetidin-2-ona e processo para a produção dos mesmos |
| TW200806623A (en) * | 2005-10-05 | 2008-02-01 | Merck & Co Inc | Anti-hypercholesterolemic compounds |
| US20100069347A1 (en) * | 2006-11-02 | 2010-03-18 | Morriello Gregori J | Heterocyclyl-substituted anti-hypercholesterolemic compounds |
| AU2007342603A1 (en) * | 2006-12-20 | 2008-07-17 | Merck Sharp & Dohme Corp. | Anti-hypercholesterolemic compounds |
-
2005
- 2005-11-23 DE DE102005055726A patent/DE102005055726A1/de not_active Withdrawn
-
2006
- 2006-11-13 JP JP2008541615A patent/JP2009516714A/ja not_active Abandoned
- 2006-11-13 EP EP06829012A patent/EP1954673A1/de not_active Withdrawn
- 2006-11-13 WO PCT/EP2006/010840 patent/WO2007059871A1/de active Application Filing
-
2008
- 2008-05-19 US US12/122,991 patent/US20080274947A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005047248A1 (en) * | 2003-11-10 | 2005-05-26 | Microbia, Inc. | 4-biarylyl-1-phenylazetidin-2-ones |
| WO2005069900A2 (en) * | 2004-01-16 | 2005-08-04 | Merck & Co., Inc. | Npc1l1 (npc3) and methods of identifying ligands thereof |
| WO2006086562A2 (en) * | 2005-02-09 | 2006-08-17 | Microbia, Inc. | Phenylazetidinone derivatives |
| WO2006116499A1 (en) * | 2005-04-26 | 2006-11-02 | Microbia, Inc. | 4-biarylyl-1-phenylazetidin-2-one glucuronide derivatives for hypercholesterolemia |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7871998B2 (en) | 2003-12-23 | 2011-01-18 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7863265B2 (en) | 2005-06-20 | 2011-01-04 | Astrazeneca Ab | 2-azetidinone derivatives and their use as cholesterol absorption inhibitors for the treatment of hyperlipidaemia |
| US7893048B2 (en) | 2005-06-22 | 2011-02-22 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7906502B2 (en) | 2005-06-22 | 2011-03-15 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7842684B2 (en) | 2006-04-27 | 2010-11-30 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2009140932A2 (en) * | 2008-05-21 | 2009-11-26 | Zentiva, K.S. | Method of producing (3r,4s)-l-(4-fluorophenyl)-3-[(3s)-3-(4-fluorophenyl)- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone |
| WO2009140932A3 (en) * | 2008-05-21 | 2010-01-14 | Zentiva, K.S. | Method of producing (3r,4s)-l-(4-fluorophenyl)-3-[(3s)-3-(4-fluorophenyl)- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2012004382A1 (en) * | 2010-07-09 | 2012-01-12 | Moehs Iberica S.L. | New method for preparing ezetimibe |
| WO2012030165A2 (ko) | 2010-08-31 | 2012-03-08 | 서울대학교산학협력단 | P P A R δ 활성물질의 태자재프로그래밍용도 |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| EP2567959A1 (de) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridin-4-Carbonsäureamid-Derivate als Kinaseinhibitoren |
| WO2014093189A1 (en) | 2012-12-10 | 2014-06-19 | Merck Sharp & Dohme Corp. | Methods of treating diabetes by administering a glucagon receptor antagonist in combination with a cholesterol absorption inhibitor |
| US11077092B2 (en) | 2012-12-10 | 2021-08-03 | Merck Sharp & Dohme Corp. | Methods of treating diabetes by administering a glucagon receptor antagonist in combination with a cholesterol absorption inhibitor |
| WO2017050990A1 (fr) * | 2015-09-25 | 2017-03-30 | Universite De Nantes | 1,4-di-(4-methylthiophenyl)-3-phtaloylazetidine-2-one et ses derives |
| FR3041641A1 (fr) * | 2015-09-25 | 2017-03-31 | Univ Nantes | 1,4-di-(4-methylthiophenyl)-3-phtaloylazetidine-2-one et ses derives |
| US10457665B2 (en) | 2015-09-25 | 2019-10-29 | Universite De Nantes | 1,4-di-(4-methylthiophenyl)-3-phtaloylazetidine-2-one and the derivatives thereof |
| CN108290868B (zh) * | 2015-09-25 | 2021-06-01 | 南特大学 | 1,4-二-(4-甲硫基苯基)-3-邻苯二甲酰氮杂环丁烷-2-酮及其衍生物 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1954673A1 (de) | 2008-08-13 |
| DE102005055726A1 (de) | 2007-08-30 |
| US20080274947A1 (en) | 2008-11-06 |
| JP2009516714A (ja) | 2009-04-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2024340B1 (de) | 4,5-diphenyl-pyrimidinyl-amino substituierte carbonsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP2024347B1 (de) | Phenylamino-benzoxazol substituierte carbonsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP2024338B1 (de) | 4,5-diphenyl-pyrimidinyl substituierte carbonsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| DE102006021872B4 (de) | 4,5-Diphenyl-pyrimidinyl-oxy oder -mercapto substituierte Carbonsäuren, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| EP2084172B1 (de) | Neue mit benzylresten substituierte 1,4-benzothiepin-1,1-dioxidderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| EP2084173B1 (de) | Neue mit fluor substituierte 1,4-benzothiepin-1,1-dioxidderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| EP2083832B1 (de) | Neue 1,4-benzothiepin-1,1-dioxidderivate mit verbesserten eigenschaften, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| EP2084143B1 (de) | Neue mit cyclohexylresten substituierte1,4-benzothiepin-1,1 -dioxidderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| EP2152711B1 (de) | 2 -heteroaryl- pyrrolo [3, 4-c]pyrrol- derivate und deren verwendung als scd inhibitoren | |
| EP2328898B1 (de) | 2-heteroaryl-pyrrolo[3,4-c]pyrrol- derivate und ihre verwendung als scd inhibitoren | |
| EP1954673A1 (de) | Hydroxy-substituierte diphenylazetidinone zur behandlung von hyperlipidämie | |
| EP1940807B1 (de) | Carbamoylbenzotriazol-derivate als inhibitoren von lipasen und phospholipasen | |
| WO2008052658A1 (de) | Neues mit piperazin-1-sulfonsäure substituiertes diphenylazetidinon mit verbesserten pharmakologischen eigenschaften | |
| DE102007063671A1 (de) | Neue kristalline Diphenylazetidinonhydrate, diese Verbindungen enthaltende Arzneimittel und deren Verwendung | |
| DE102007005045B4 (de) | Phenothiazin Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006829012 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008541615 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006829012 Country of ref document: EP |