WO2008016811A2 - Aminopiperidines and realted compounds - Google Patents
Aminopiperidines and realted compounds Download PDFInfo
- Publication number
- WO2008016811A2 WO2008016811A2 PCT/US2007/074282 US2007074282W WO2008016811A2 WO 2008016811 A2 WO2008016811 A2 WO 2008016811A2 US 2007074282 W US2007074282 W US 2007074282W WO 2008016811 A2 WO2008016811 A2 WO 2008016811A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- mono
- hydrate
- salt
- Prior art date
Links
- 0 CCC(C)CCC(C)(CC(C)CC)N** Chemical compound CCC(C)CCC(C)(CC(C)CC)N** 0.000 description 24
- VCNBYGUQLNAILR-UHFFFAOYSA-N CC(C(C=C1)=C(C)C(C)C1OCCCCN(C)C)N(C(CC1)C2)C1C2NC(C1=CC(C2[IH]CC2)NC=C1)=O Chemical compound CC(C(C=C1)=C(C)C(C)C1OCCCCN(C)C)N(C(CC1)C2)C1C2NC(C1=CC(C2[IH]CC2)NC=C1)=O VCNBYGUQLNAILR-UHFFFAOYSA-N 0.000 description 1
- REKSPJBZUYHEAV-BZSJEYESSA-N CC(c(c(C)c1C)ccc1OC)N(CC1)C[C@@H]1NC(c(ncc(F)c1)c1F)=O Chemical compound CC(c(c(C)c1C)ccc1OC)N(CC1)C[C@@H]1NC(c(ncc(F)c1)c1F)=O REKSPJBZUYHEAV-BZSJEYESSA-N 0.000 description 1
- ONKQCFUCYKPQFU-OMOCHNIRSA-N CC(c(c(C)c1C)ccc1OC)N(CC1)C[C@@H]1NC(c1cc(Cl)ccn1)=O Chemical compound CC(c(c(C)c1C)ccc1OC)N(CC1)C[C@@H]1NC(c1cc(Cl)ccn1)=O ONKQCFUCYKPQFU-OMOCHNIRSA-N 0.000 description 1
- HYDZGOJRMUMNLW-XLLDNJMBSA-N CC(c(c(C)c1C)ccc1OCCN1[C@@H](O)OCC1)[N](CC1)(C1C1)C=C[C@@H]1NC(c(cc1Cl)ccc1F)O Chemical compound CC(c(c(C)c1C)ccc1OCCN1[C@@H](O)OCC1)[N](CC1)(C1C1)C=C[C@@H]1NC(c(cc1Cl)ccc1F)O HYDZGOJRMUMNLW-XLLDNJMBSA-N 0.000 description 1
- XTXZZBGSEUVDMV-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OC)N(C(CC1)C2)C1CC2N Chemical compound CC(c(cc1)c(C)c(C)c1OC)N(C(CC1)C2)C1CC2N XTXZZBGSEUVDMV-UHFFFAOYSA-N 0.000 description 1
- MQDLAFYPMXXCMH-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OC)N(CC1)CCC1N(CCOC)C(c(cc1Cl)ccc1F)=O Chemical compound CC(c(cc1)c(C)c(C)c1OC)N(CC1)CCC1N(CCOC)C(c(cc1Cl)ccc1F)=O MQDLAFYPMXXCMH-UHFFFAOYSA-N 0.000 description 1
- WHBFLAOPYNDZNR-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OC)N1CC(CN)CC1 Chemical compound CC(c(cc1)c(C)c(C)c1OC)N1CC(CN)CC1 WHBFLAOPYNDZNR-UHFFFAOYSA-N 0.000 description 1
- CCHROQXHLIGNKG-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OC)N1CC(CNC(c(cc2)ccc2SC)=O)CCC1 Chemical compound CC(c(cc1)c(C)c(C)c1OC)N1CC(CNC(c(cc2)ccc2SC)=O)CCC1 CCHROQXHLIGNKG-UHFFFAOYSA-N 0.000 description 1
- ASKYODBWFGOMQC-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OCC1OCCNC1)N(C(C1)COC2)C2C1NC(c1cc(F)ccc1)=O Chemical compound CC(c(cc1)c(C)c(C)c1OCC1OCCNC1)N(C(C1)COC2)C2C1NC(c1cc(F)ccc1)=O ASKYODBWFGOMQC-UHFFFAOYSA-N 0.000 description 1
- UPGZWIKFZQVKGU-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OCCCCCN(C)C)N(C(CC1)C2)C1CC2NC(c1cc(Cl)cc(Cl)c1)=O Chemical compound CC(c(cc1)c(C)c(C)c1OCCCCCN(C)C)N(C(CC1)C2)C1CC2NC(c1cc(Cl)cc(Cl)c1)=O UPGZWIKFZQVKGU-UHFFFAOYSA-N 0.000 description 1
- SUNNPEVVEDEAMY-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OCCCCN(C)C)N1C(CC(C2)NC(C3CCCCC3)=O)C22C1CC2 Chemical compound CC(c(cc1)c(C)c(C)c1OCCCCN(C)C)N1C(CC(C2)NC(C3CCCCC3)=O)C22C1CC2 SUNNPEVVEDEAMY-UHFFFAOYSA-N 0.000 description 1
- KAAVYCPLMQQPRB-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OCCCCN(C)C)N1C(CC(C2)NC=O)C22C1CC2 Chemical compound CC(c(cc1)c(C)c(C)c1OCCCCN(C)C)N1C(CC(C2)NC=O)C22C1CC2 KAAVYCPLMQQPRB-UHFFFAOYSA-N 0.000 description 1
- DSEFQLZZMSKMDN-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OCC[O]=C)N(CC1)CCC1NC(CSC)=O Chemical compound CC(c(cc1)c(C)c(C)c1OCC[O]=C)N(CC1)CCC1NC(CSC)=O DSEFQLZZMSKMDN-UHFFFAOYSA-N 0.000 description 1
- BGWGWJAIYKDTOT-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OCCc1c(C)[s]c(C)n1)N(C(CC1)C2)C1CC2NC(c(cc1Cl)ccc1F)=O Chemical compound CC(c(cc1)c(C)c(C)c1OCCc1c(C)[s]c(C)n1)N(C(CC1)C2)C1CC2NC(c(cc1Cl)ccc1F)=O BGWGWJAIYKDTOT-UHFFFAOYSA-N 0.000 description 1
- MVTZNOUNVDTNQW-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OCCc1c(C)[s]c(N(C)C)n1)N(C(CC1)C2)C1CC2NC(c(cc1)cc(Cl)c1F)=O Chemical compound CC(c(cc1)c(C)c(C)c1OCCc1c(C)[s]c(N(C)C)n1)N(C(CC1)C2)C1CC2NC(c(cc1)cc(Cl)c1F)=O MVTZNOUNVDTNQW-UHFFFAOYSA-N 0.000 description 1
- JRSYQVFJCAYLON-UHFFFAOYSA-N CC(c(cc1)c(C)c(C)c1OCCc1ccn[n]1C)N(C(CC1)C2)C1C(C1)C21NC(c(cc1Cl)ccc1F)=O Chemical compound CC(c(cc1)c(C)c(C)c1OCCc1ccn[n]1C)N(C(CC1)C2)C1C(C1)C21NC(c(cc1Cl)ccc1F)=O JRSYQVFJCAYLON-UHFFFAOYSA-N 0.000 description 1
- ASYPTQNEKLBTSH-PMRCOVRLSA-N CC(c(cc1)c(C)c(C)c1OC[C@@H](CC1)NC1=O)NC(CC[C@@H](C)C1)C[C@H]1NC1O[C@H]1C1CCCC1 Chemical compound CC(c(cc1)c(C)c(C)c1OC[C@@H](CC1)NC1=O)NC(CC[C@@H](C)C1)C[C@H]1NC1O[C@H]1C1CCCC1 ASYPTQNEKLBTSH-PMRCOVRLSA-N 0.000 description 1
- BSOMZJGHIJTVHJ-UHFFFAOYSA-N CCC(c(cc1)c(C)c(C)c1OC)N1CC(CNC(c(cccc2)c2OC)=O)CCC1 Chemical compound CCC(c(cc1)c(C)c(C)c1OC)N1CC(CNC(c(cccc2)c2OC)=O)CCC1 BSOMZJGHIJTVHJ-UHFFFAOYSA-N 0.000 description 1
- NQBKAMBRPQEBOX-UHFFFAOYSA-N CCNC(c1cc(C)c(C(C)N(CC2)CCC2NC(c(cc2Cl)ccc2F)=O)cc1)=O Chemical compound CCNC(c1cc(C)c(C(C)N(CC2)CCC2NC(c(cc2Cl)ccc2F)=O)cc1)=O NQBKAMBRPQEBOX-UHFFFAOYSA-N 0.000 description 1
- FJWROMVHFIGSHH-UHFFFAOYSA-N CCc1c(C)c(C(C)N2CC(CNC(c(cc3)ccc3F)=O)C2)ccc1OCCCCO Chemical compound CCc1c(C)c(C(C)N2CC(CNC(c(cc3)ccc3F)=O)C2)ccc1OCCCCO FJWROMVHFIGSHH-UHFFFAOYSA-N 0.000 description 1
- ARMPZWIXJACTSN-VIQWUECVSA-N COc1cc(O)c(CN2C(C3)COCCC2C[C@@H]3NC(c2cc(F)ccc2)=O)c(OC)c1 Chemical compound COc1cc(O)c(CN2C(C3)COCCC2C[C@@H]3NC(c2cc(F)ccc2)=O)c(OC)c1 ARMPZWIXJACTSN-VIQWUECVSA-N 0.000 description 1
- URWWRSQNQXKLPF-MSNNAKFMSA-N C[C@@H](C1C=C=CC)C=CC(C)=C1F Chemical compound C[C@@H](C1C=C=CC)C=CC(C)=C1F URWWRSQNQXKLPF-MSNNAKFMSA-N 0.000 description 1
- CXYXNSXRGVUUAJ-SWOOVLCTSA-N C[C@@H](COc1c(C)c(C)c(C(C)N(C(CC2)C3)C2CC3NC(c(cc2Cl)ccc2F)=O)cc1)O Chemical compound C[C@@H](COc1c(C)c(C)c(C(C)N(C(CC2)C3)C2CC3NC(c(cc2Cl)ccc2F)=O)cc1)O CXYXNSXRGVUUAJ-SWOOVLCTSA-N 0.000 description 1
- OMBYECRTIAHEMC-UHFFFAOYSA-N Cc(cc1)c(C)cc1C(NCC1N(Cc(cc2)c(C)c(C)c2OC)CCCC1)=O Chemical compound Cc(cc1)c(C)cc1C(NCC1N(Cc(cc2)c(C)c(C)c2OC)CCCC1)=O OMBYECRTIAHEMC-UHFFFAOYSA-N 0.000 description 1
- KILMDVZWJYPXIX-UHFFFAOYSA-N Cc1c(CN(C(CC2)C3)C2CC3NC(C(C=CC2F)=CC2Cl)=O)ccc(OCCCN2CCOCC2)c1C Chemical compound Cc1c(CN(C(CC2)C3)C2CC3NC(C(C=CC2F)=CC2Cl)=O)ccc(OCCCN2CCOCC2)c1C KILMDVZWJYPXIX-UHFFFAOYSA-N 0.000 description 1
- JAQAOVTZIPHZLV-UHFFFAOYSA-N Cc1c(CN(C(CC2)C3)C2CC3NC(Cc(ccc(Cl)c2)c2Cl)=O)ccc(OCCOC)c1C Chemical compound Cc1c(CN(C(CC2)C3)C2CC3NC(Cc(ccc(Cl)c2)c2Cl)=O)ccc(OCCOC)c1C JAQAOVTZIPHZLV-UHFFFAOYSA-N 0.000 description 1
- ACXHHJQFSAMFLM-ZCJYOONXSA-N Cc1c(CN(CCC2)[C@@](C3)(C3NC(Cc(cc3Cl)ccc3Cl)=O)C2=[IH])ccc(OCCOC)c1C Chemical compound Cc1c(CN(CCC2)[C@@](C3)(C3NC(Cc(cc3Cl)ccc3Cl)=O)C2=[IH])ccc(OCCOC)c1C ACXHHJQFSAMFLM-ZCJYOONXSA-N 0.000 description 1
- HMBTZIHBDKFDDO-UHFFFAOYSA-N Cc1c(CNC(CC2)(C3)C2CC3NC(Cc(cc2)ccc2Br)=O)ccc(OCCOC)c1C Chemical compound Cc1c(CNC(CC2)(C3)C2CC3NC(Cc(cc2)ccc2Br)=O)ccc(OCCOC)c1C HMBTZIHBDKFDDO-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D411/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen and sulfur atoms as the only ring hetero atoms
- C07D411/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen and sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D411/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen and sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
- C07D451/04—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof with hetero atoms directly attached in position 3 of the 8-azabicyclo [3.2.1] octane or in position 7 of the 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring system
Definitions
- This invention relates generally to aminopiperidines and related compounds.
- the invention further relates to the use of such compounds for treating a variety of metabolic, eating and sexual disorders, and as probes for the detection and localization of melanin concentrating hormone receptors.
- MCH Melanin concentrating hormone
- Agents capable of modulating MCH receptor activity are highly desirable for the treatment of a variety of diseases and disorders, including obesity, metabolic syndrome, eating disorders (e.g., bulimia and anorexia), sexual disorders (e.g., anorgasmic or psychogenic impotence) and metabolic disorders, such as diabetes.
- Small molecule, non-peptide antagonists of MCH receptors would be of particular value for such therapies.
- the present invention fulfills this need, and provides further related advantages.
- R A is 6- to IO-membered aryi, 5- to I O-membered heteroaryl, Q-Csalkyl, Cj-Cehaloaikyl, (C 3 - Cgcycloalkyl)Co-Gia]kyl or (5- to 7-membered heterocycloalkyl)Co-QalkyI, each of which is optionally substituted and each of which is preferably substituted with from 0 to 4 substituents independently chosen from R B ;
- Each R B is independently:
- U and T are independently N or CRy; P is N or CRi 5 ; Q is N or CR 9 ; Ri is cyano, nitro, halogen or a group of the formula -L-M, wherein:
- M is hydrogen, Q-CgalkyL C 2 -C 8 alkenyl, C 2 -C 8 aikynyl, C 2 -C 8 alkyl ether, mono- or di-(Q-
- R is taken together with R 9 to form a fused C 5 -C 3 cycloalkyi or 5- to 8-membered heterocycloalkyl; in certain embodiments, R 1 is not H;
- R 2 represents from 0 to 4 substituents independently chosen from Q -C h alky], C r C 4 haloalkyl and oxo; or two R 2 groups are taken together to form a bridge (e.g., methylene, ethylene, propylene or -CH 2 -O-CH 2 -);
- R 3 is hydrogen, C]-C 4 aikyi or Ci ⁇ hal ⁇ alkyl;
- R 4 is:
- Each R 7 , R 9 and R 15 is independently hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH. aminocarbonyl. aminosulfonyi, CrQalkyl. C 2 -Qalkenyl, C 2 -C 6 alkynyl, C]-C 6 alkoxy, C r
- R 9 is taken together with R ; to form a fused cycloalkyl or heterocycloalkyl; and R I G is hydrogen, Q-Cgalkyl, C ⁇ -Cealkenyl or C 2 -C6alkynyL
- R I G is hydrogen, Q-Cgalkyl, C ⁇ -Cealkenyl or C 2 -C6alkynyL
- Rs is hydrogen, Q-Cgalkyl, C 2 -Cgaikenyl, Cj-CgaJkynyl.
- R 9 and R !5 are independently halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl, aminosulfonyl, CrQalkyl, C 2 -Qalkenyl, C 2 -Qalkynyl. Cj-Qalkoxy, CpCealkanoyl, C ⁇ - C 6 alkanone, Ci-C 6 alkanoyloxy, C 2 -C 6 alky ⁇ ether, C]-C 6 alkoxycarbonyl, Ci-Csalkylthio. Q- Csalkylsulfonyl. mono- or di-(C ⁇ -C6alkyl)aminoC 0 -C 4 alkyl, mono- or di-(Ci-
- R 2 represents from 1 to 4 substituents independently chosen from Q ⁇ alkyl, Cr
- P is N, CH or CR 15 ;
- Ri is a group of the formula -L-M, wherein: L is a single covalent bond, O, C(O) 8 OC(O), C(O)O, OC(O)O, S(0) w , N(R x ),
- each R x is independently hydrogen, C,-C 6 alkyl, C 2 -C 6 aikenyl.
- C 2 -C 6 alkynyl or C r C 6 haloalkyl and w is independently selected at each occurrence from O, 1 and 2;
- M is hydrogen, Ci-C 3 alkyi, Ci-Cgalkenyl, C ⁇ -Cgalkynyl, Ca-Cgalkyl ether, mono- or di-(C r C 8 aiky])aminoC 0 -C 6 alkyl, (C 3 -C 7 cycloalkyi)Co-C 6 aikyI or (5- to 10-membered heterocycle)Co-C 6 aikyl, each of which is substituted with from O to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbo ⁇ yl, imino, aminosulfonyl, -COOH, cyanoimido, CrQalkyl, Ci-Cghaloalkyl, Ci-C f ialkoxy, C r
- R] is taken together with R 9 to form a fused Cs-Cgcycloalkyl or 5- to 8-membered heterocyc loal ky i ; such that R] is not hydrogen;
- 5 are independently halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl, aminosulfonyl.
- Cj-Cealkyi C 2 -C 6 alkenyl, C ⁇ -C ⁇ alkynyl, C]-C 6 alkanoyl, Cs-Qalkanone, C;- C 6 a ⁇ kanoy!oxy, C 2 -C 5 alkyi ether, C r C 6 alkoxycarbonyl, C]-C e alkylthio ; Cs-C ⁇ alkylsulfonyl, mono- or di-(Ci-C ⁇ alkyl)ammoCo-C 4 alkyl, mono- or di»(C r C ⁇ a!kyl)aminocarbonylCo-C 4 afkyl, mono- or di-fCrQalkylJaminosuifo ⁇ yiCo-Cjafkyl, CpCehaloalkyl, Ci-Cehaloalkoxy, Cp C ⁇ hydroxyalkyl, Ci-C 6 aminoalkyl, Ci-C 6 cyanoalkyl, or mono
- Formula VII Formula VIII or are a pharmaceutically acceptable salt, solvate or esters of such a compound.
- Variables within Formulas V, VI 3 VIl and VIII are generally as described for Formula I.
- Formula IX are a pharmaceutically acceptable salt, solvate or esters of such a compound. Variables within
- Formula TX are generally as described for Formula I.
- aminopiperidines and related compounds provided herein are MCH receptor modulators and exhibit a K 1 of no greater than 1 micromolar, 500 nanomolar, 100 nanomolar, or 10 nanomolar in a MCH receptor binding assay and/or have an ECj 0 or IC 5 O value of no greater than 1 micromolar, 500 nanomolar, 100 nanomolar, or 10 nanomolar in an assay for determining MCH receptor agonist or antagonist activity.
- aminopiperidines and related compounds provided herein are labeled with a detectable marker (e.g., radiolabeled or fluorescein conjugated).
- a detectable marker e.g., radiolabeled or fluorescein conjugated.
- the present invention further provides, within other aspects, pharmaceutical compositions comprising at least one aminopiperidine or related compound provided herein in combination with a physiologically acceptable carrier or excipient.
- a pharmaceutical composition provided herein may further comprise one or more additional active agents (i.e., drugs).
- compositions provided herein may be formulated, for example, as an injectable fluid, an aerosol, a cream, an oral liquid, a tablet, a gel, a pill, a capsule, a syrup or a transdermal patch.
- Methods are further provided for modulating binding of ligand (e.g., MCH) to cellular MCH receptor, comprising contacting cells expressing MCH receptor with a MCH receptor modulator as described above, in an amount that would be sufficient to detectab ⁇ y modulate MCH binding to MCH receptor in vitro.
- the cells may, but need not, be present in a human nor non-human animal. 5 Tn other aspects, methods are provided for modulating binding of ligand (e.g., MCH) to
- MCH receptor in vitro comprising contacting MCH receptor with a MCH receptor modulator as described above, in an amount sufficient to detectably modulate MCH binding to MCH receptor.
- the present invention provides methods for modulating the signal- transducing activity of MCH receptor in a cell, comprising contacting a cell expressing MCH I O receptor, either in vivo or in vitro, with a MCH receptor modulator as described above, under conditions and in an amount that is sufficient to detectably alter the electrophysiology of the cell.
- the MCH receptor is a MCHlR.
- the present invention further provides, within other aspects, methods for treating a disease or disorder associated with MCH receptor activation, comprising administering to a patient in need 5 of such treatment a therapeutically effective amount of a MCH receptor modulator as described above.
- Such diseases and disorders include, for example, obesity, metabolic syndrome, eating disorders (e.g., bulimia nervosa), sexual disorders, diabetes, heart disease and stroke.
- the MCH receptor modulator may be administered oralfy, or via another means such as intranasal Iy, intravenously or topically.
- the patient is a human, companion animal0 (e.g., dog or cat) or livestock.
- MCH receptor modulator and administering to the patient an effective amount of a MCH receptor5 modulator as described above.
- Methods are provided, within other aspects, for determining the presence or absence of MCH receptor in a sample, comprising: (i) contacting a sample with a compound as described above under conditions that permit binding of the compound to MCH receptor; and (ii) detecting a level of the compound bound to MCH receptor.
- the compound is radiolabeled,0 and the step of detection comprises: (i) separating unbound compound from bound compound; and (ii) determining an amount of bound compound in the sample. Detection may be achieved, for example, using autoradiography.
- Representative samples include, for example, tissue sections.
- Packaged pharmaceutical preparations comprising: (a) a pharmaceutical composition as described above in a container; and (b) instructions for using the composition to treat5 a patient suffering from or at risk for developing a disease or disorder associated with MCH receptor activation.
- methods for preparing the compounds disclosed herein, including the intermediates are also provided herein.
- the present invention provides aminopiperidines and related compounds of
- MCH receptor modulators that may be used in vitro or in vivo, to inhibit MCH binding to MCH receptors, activate MCH receptors, or to otherwise modulate MCH receptor activity in a variety of contexts, as discussed in further detail below.
- isotopes of hydrogen include tritium and deuterium and isotopes of carbon include 11 C, 13 C and ]A C.
- Certain compounds are described herein using a genera! formula that includes variables (e.g., X, V, R 3 ). Unless otherwise specified, each variable within such a formula is defined independently of any other variable, and any variable that occurs more than one time in a formula is defined independently at each occurrence. In general, the variables may have any definition described herein that results in a stable compound.
- aminopiperidines and related compounds refers to any compound that satisfies Formula I, or other Formula provided herein, or is a pharmaceutically acceptable salt, solvate (e.g., hydrate) or ester of such a compound. Certain aminopiperidines and related compounds further satisfy one or more additional formulas provided herein.
- a "pharmaceutically acceptable salt' 1 of a compound recited herein is an acid or base salt that is suitable for use in contact with the tissues of human beings or animals without excessive toxicity or carcinogenicity, and preferably without irritation, allergic response, or other problem or complication.
- Such salts include mineral and organic acid salts of basic residues such as amines, as well as alkali or organic salts of acidic residues such as carboxylic acids.
- Specific pharmaceutically acceptable anions for use in salt formation include, but are not limited to, acetate, 2- acetoxybenzoate, ascorbate, benzoate, bicarbonate, bromide, calcium edetate, carbonate, chloride, citrate, dihydrochloride, diphosphate, ditartrate, edetate, estolate (ethylsuccinate). formate, fumarate, gluceptate.
- gluconate glutamate, glycolate, glycollylarsanilate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroiodide, hydroxymaieate, hydroxynaphthoate, iodide, isethionate, lactate, lactobionate, malate, maleate, mandelate, methylbroraide, methylnitrate, methylsulfate, mucate, napsylate, nitrate, pamoate, pantothenate, phenylacetate, phosphate, polygalacturonate, propionate, salicylate, stearate, subacetate, succinate, sulfamate, sulfanilate, sulfate, sulfonates including besylate (benzenesuifonate).
- carnsylate (camphorsulfonate), edisylate (ethane- 1 ,2- disulfonate), esylate (ethanesulfonate) 2-hydroxy ethyl sulfonate, mesylate (methanesulfonate), Inflate (trifluoromethanesulfonate) and tosylale (p-toluenesulfonate), tannate, tartrate, teoclate and trieth iodide.
- pharmaceutically acceptable cations for use in salt formation include, but are not limited to ammonium, benzathine, chloroprocaine, choline, diethanolamine, ethylenediami ⁇ e, meglumine, procaine, and metals such as aluminum, calcium, lithium, magnesium, potassium, sodium and zinc.
- a pharmaceutically acceptable acid or base salt can be synthesized from a parent compound that contains a basic or acidic moiety by any conventional chemical method.
- such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, the use of nonaqueous media, such as ether, ethyl acetate, ethanol, methanol, isopropanol or acetonitrile, is preferred.
- nonaqueous media such as ether, ethyl acetate, ethanol, methanol, isopropanol or acetonitrile
- each compound of Formula I may, but need not, be formulated as a solvate (e.g., a hydrate) or a non-covalent complex.
- the various crystal fo ⁇ ns and polymorphs are within the scope of the present invention.
- prodrugs of the aminopiperidines and related compounds provided herein.
- a "prodrug” is a compound that may not fully satisfy the structural requirements of the compounds provided herein, but is modified in vivo, following administration to a patient, to produce an aminopiperidine or related compound.
- a prodrug may be an acylated derivative of a compound as provided herein.
- Prodrugs include compounds wherein hydroxy, amine or sulfhydryi groups are bonded to any group that, when administered to a mammalian subject, cleaves to form a free hydroxy, amino or sulfhydryl group, respectively.
- prodrugs include, but are not limited to, acetate, formate, phosphate and benzoate derivatives of alcohol and amine functional groups within the compounds provided herein.
- Prodrugs of the compounds provided herein may be prepared by modifying functional groups present in the compounds in such a way that the modifications are cleaved in vivo to yield the parent compounds.
- alkyl refers to a straight or branched chain saturated aliphatic hydrocarbon. Alkyl groups include groups having from 1 to 8 carbon atoms (Ci-Qalkyl), from 1 to
- Ci-C 6 alkyl and from 1 to 4 carbon atoms (Cj-C 4 alkyl), such as methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl, tert-butyi, pentyl, 2-pentyl, isopentyl, neopentyl, hexyl, 2- hexyl, 3-hexyl and 3-methylpentyl.
- C 0 -C n a3kyl refers to a single covalent bond (C 0 ) or an alky!
- CVCealkyi refers to a single covalent bond or a Cj-Qalkyl group. In some instances, a substituenl of an alky! group is specifically indicated.
- Ci-Cehydroxyalkyl refers to a CrCgalkyl group that has at least one hydroxy substituent
- Ci-C 6 aminoalkyl refers to a C r C 6 alkyI group that has at least one amino substituent
- Ci-C ⁇ Cyanoalkyi refers to a CpCsalkyl group that has at least one cyano (C ⁇ N) substituent.
- Alkylene refers to a divalent alkyi group, as defined above.
- Co-C 4 alkyle ⁇ e is a single covalent bond or an alkylene group having from 1 to 4 carbon atoms.
- Alkenyl refers to straight or branched chain alkene groups, which comprise at least one unsaturated carbon-carbon double bond. Alkenyl groups include, for example, C 2 -C 8 alkenyl and
- alkenyi groups which have from 2 to 8 or from 2 to 6 carbon atoms, respectively ⁇ e.g., ethenyl, allyl or isopropenyl).
- alkynyl refers to straight or branched chain alkyne groups, which have one or more unsaturated carbon-carbon bonds, at least one of which is a triple bond.
- Aikynyl groups include, for example, Cj-Cgalkynyl and Ci-Cgalkynyl groups, which have from 2 to 8, or from 2 to 6 carbon atoms, respectively.
- a “cycloalkyl” is a group that comprises one or more saturated and/or partially saturated rings in which ail ring members are carbon, such as cyclopropyl, cyciobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, adamantyl. decahydr ⁇ -naphthalenyl, octahydro-indenyl, and partially saturated variants of the foregoing, such as cyclohexenyl.
- Certain cycloalkyl groups are Ci -C 7 cycloalkyl, in which the group contains from 3 to 7 ring members.
- a "cycloalkyiCo-C n alkyi” is a cycloalkyl group linked via a single covalent bond or a C r C n alkylene group (e.g., (C 3 - C 7 cycloalkyl)C 0 -C,alkyl).
- alkoxy is meant an alky I group as described above attached via an oxygen bridge.
- Alkoxy groups include Cj-C ⁇ alkoxy and C
- Methoxy, ethoxy. propoxy, isopropoxy, n-butoxy, sec- butoxy. ter/-butoxy, n-pentoxy, 2-pentoxy, 3-pentoxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy. and 3-methyIpentoxy are representative alkoxy groups.
- alkylthio refers to an alkyl group as described above attached via a sulfur bridge.
- Alkylsulfonyl refers to groups of the formula - ⁇ S ⁇ 2 )-aikyL in which the sulfur atom is the point of attachment.
- Alkylsulfonyl groups include Ci-C 6 alkylsulfonyl and Ci-C 4 alkylsuifonyl groups, which have from 1 to 6 or from 1 to 4 carbon atoms, respectively.
- Methylsulfonyl is one representative alkylsulfonyl group.
- Alkanoyl groups have the indicated number of carbon atoms, with the carbon of the keto group being included in the numbered carbon atoms.
- - Alkanoyl groups include, for example, C ⁇ -Cealkanoyl, which have from 2 to 6 carbon atoms.
- Alkyl ether refers to a linear or branched ether substituent (i.e., an alkyl group that is substituted with an alkoxy group). Such groups include C 2 -C 3 alkyi ether and C 2 -C 6 alkyl ether. A C 2 alkyl ether group has the structure -CH 2 -O-CH 3 .
- Alkoxycarbonyl groups include, for example, Ci-Ce and C]-C 4 alkoxycarbonyl groups, which have from 1 to 6 or from 1 to 4 carbon atoms, respectively, in the alkyl portion of the group (i.e., the carbon of the keto bridge is not included in the indicated number of carbon atoms).
- Alkanoylamino groups include, for example, C 2 -C 6 alkanoylamino groups, which have from 2 to 6 carbon atoms.
- Alkylamino refers to a secondary or tertiary amine having the general structure -NH- alkyl or -N ⁇ alkyl)(alkyl), wherein each "alkyl” is selected independently from alkyl, cycloalkyl and
- (cycioaikyl)alkyl groups include, for example, mono- and di-(C]-CgalkyI)amino groups, as well as mono- and di-(C
- Alkylaminoalkyl refers to an aikylamino group linked via an alkylene group (i.e., a group having the general structure -alkylene-NH-alkyl or -alkyIene-N(aIkyl)(alkyI), in which each alkyi is selected independently from alkyl, cycloalkyl and (cycloalkyl)alkyl groups).
- Alkylaminoalkyl groups include, for example, mono- and di-(Ci-Cgalkyl)aminoCrCga!kyl, mono- and di-(Cj- C 6 atkyl)ammoCj-C 6 alkyl and mono- and dHCj-C ⁇ alkyOaminoCi-Qalkyl.
- "Mono- or di-(C r C 6 alkyl)aminoC 0 -C 6 alkyl” refers to a mono- or di-(C r C 6 alky!)amino group linked via a single covalent bond or a Ci-Cealkylene group.
- alkyl as used in the terms “alkylamino” and “alkylaminoalkyl” differs from the definition of "alkyl” used for all other alkyl-containing groups, in the inclusion of cyc ⁇ oalkyi and (cycloalkyl)a!kyl groups (e.g., tC 3 -C 7 cycloaikyl)C 0 -C 6 alkyl).
- “Mono- or di-(C r Cgalkyl)aminocarbonyi” is an aminocarbonyl group in which one or both of the hydrogen atoms is replaced with Ci-Cgalkyi. If both hydrogen atoms are so replaced, the alkyl groups may be the same or different.
- “Mono- or di-(CrC 6 a!kyl)aminocarbonylC 0 -C 4 alkyl is a mono- or di-(C r
- Aminosulfonyi refers to groups of the formula "-(SOi)-NH 2 , in which the sulfur atom is the point of attachment.
- the term “mono- or di-(C]-C ⁇ alkyl)aminosulfonyl” refers to groups that satisfy the formula - ⁇ SOi)--NR 2 , in which the sulfur atom is the point of attachment, and in which one R is C r C n alkyI and the other R is hydrogen or an independently chosen Cj-C ⁇ alkyl.
- “Mono- or di-(Ci-C 6 alkyl)aminosulfonylCo-C 4 alkyl is a mono- or di-(C]-C 6 alkyl)aminosulfonyl group that is attached via a single covalent bond or a Q-C ⁇ alkyiene group.
- halogen refers to fluorine, chlorine, bromine or iodine.
- a “haioalkyl” is an alkyl group that is substituted with 1 or more independently chosen halogens ⁇ e.g., "CrQhaloalkyl” groups have from 1 to 6 carbon atoms, each of which is optionally substituted with 1 or more halogens). Examples of haloalkyl groups include, but are not limited to.
- Typical haioalkyl groups are trifluoromethy! and difluoromethyl.
- haloalkoxy refers to a haloalkyi group as defined above attached via an oxygen bridge.
- C r C 6 haIoalkoxy have 1 to 6 carbon atoms.
- a dash (“-") that is not between two letters or symbols is used to indicate a point of attachment for a substituent.
- -CONH 2 is attached through the carbon atom.
- a “carbocycle” or “carbocyclic group” comprises at least one ring formed entirely by carbon-carbon bonds (referred to herein as a carbocyclic ring), and does not contain a heterocycle. Unless otherwise specified, each ring within a carbocycle may be independently saturated, partially saturated or aromatic, and is optionally substituted as indicated.
- a carbocycle generally has from 1 to 3 fused, pendant or spiro rings; carbocycies within certain embodiments have one ring or two fused rings. Typically, each ring contains from 3 to 8 ring members ⁇ i.e., C 3 -C 5 ); C 5 -C 7 rings are recited in certain embodiments.
- Carbocycies comprising fused, pendant or spiro rings typically contain from 9 to 14 ring members. Certain carbocycies are C 4 -C] 0 (i.e., contain from 4 to 10 ring members and I or two rings). Certain representative carbocycies are cycloalkyl as described above. Other carbocycies are aiyl (i.e., contain at least one aromatic carbocyclic ring, with or without one or more additional aromatic and/or cycloalkyl rings).
- Such aryl carbocycies include, for example, phenyl, naphthyl (e.g., 1-naphthyl and 2-naphthyl), biphenyl, fluorenyl, indanyl and 1 ,2,3,4- tetrahydro-naphthyl.
- preferred carbocycles are carbocycles having a single ring, such as phenyl and 3- to 7-membered cycloalkyl groups (C 3 -C7cycloalkyl).
- aryl indicates aromatic groups containing only carbon in the aromatic ring or rings. Such aromatic groups may be further substituted with carbon and/or non- carbon atoms or groups. Typical aryl groups contain 1 or 2 separate, fused, or pendant rings and from 6 to about 12 ring atoms, without heteroatoms as ring members. 6- to 10-membered aryl groups include phenyl, naphthyl and phenyl groups that are fused to a 5 to 7-membered saturated or partially saturated ring that optionally contains 1 or 2 heteroatoms independently chosen from N, O and S (e.g., 3,4-methyIenedioxy-phenyl).
- arylalkyl refers to an aryl group linked via an alkylene bridge.
- phenylCo-C ⁇ alkyl indicates a phenyl group that is attached via a single covalent bond ( ⁇ henylC o alkyi) or attached through an alkylene group having 1 or 2 carbon atoms.
- an aryl group may be attached through other linker groups; such groups include, for example, arylCV C 6 alkanoylamino and arylalkoxy groups, in which the aryl is attached via the indicated linker group.
- a “heterocycle” or “heterocyclic group” has from 1 to 3 fused, pendant or spiro rings, at least one of which is a heterocyclic ring (i.e., one or more ring atoms is a heteroatom independently chosen from O, S and N, with the remaining ring atoms being carbon). Additional rings, if present, may be heterocyclic or carbocyclic. Typically, a heterocyclic ring comprises 1, 2, 3 or 4 heteroatoms; within certain embodiments each heterocyclic ring has 1 or 2 heteroatoms per ring.
- Each heterocyclic ring generally contains from 3 to 8 ring members (rings having from 4 or 5 to 7 ring members are recited in certain embodiments) and heterocycles comprising fused, pendant or spiro rings typically contain from 9 to 14 ring members.
- Certain heterocycles comprise a sulfur atom as a ring member; in certain embodiments, the sulfur atom is oxidized to SO or SO 2 .
- Heterocycles may be optionally substituted with a variety of substituents, as indicated.
- a heterocycle may be a heterocycloalkyl group (i.e., each ring is saturated or partially saturated) or a heteroaryl group (i.e., at least one heterocyclic ring within the group is aromatic), such as a 5- to 10-membered heteroaryl (which may be monocyclic or bicyclic) or a 6- membered heteroaryl (e.g., pyridyl or pyrimidyl).
- N-linked heterocyclic groups are linked via a component nitrogen atom.
- 4-to 7-membered heterocycloalkyl groups include, for example, piperidinyl, piperazinyl.
- aromatic heterocycles include azocinyl, pyridyl, pyrimidyl, imidazolyl and tetrazolyl.
- preferred heterocycles are 5- to 7-membered heterocycles having a single saturated, partially unsaturated or aromatic heterocyclic ring with 5 to 7 ring members, 1 or 2 ring members independently chosen from N, O and S, with remaining ring members being carbon.
- heterocycles are attached via an indicated linker group (e.g., (heterocycle)alkyl, (heterocycle)alkoxy and (heterocycle)alkylamino groups).
- the heterocycle is covalently bound to the indicated linker group, each of which carries the definition set forth above.
- a (4- to 10-membered heterocycle)Co-C 2 alkyl is a heterocyclic group, which may be saturated, partially saturated or aromatic, that contains from 4 to 10 ring members and that is linked via a single covalent bond or a methylene or ethylene linker.
- heteroary indicates a monocyclic, bicyclic or tricyclic ring system that comprises at least one 5- or 6-membered heterocyclic aromatic ring that contains from 1 to 4 (preferably from 1 to 3) heteroatoms independently chosen from N, O and S, with remaining ring atoms being carbon. If the total number of S and O atoms in the heteroaryl group exceeds 1, these heteroatoms are not adjacent to one another. It is generally preferred that the total number of S and O atoms in the heteroaryl group is not more than 2; in certain embodiments, the total number of S and O atoms in the aromatic heterocycle is not more than 1.
- heteroaryl groups include, but are not limited to, oxazolyl, pyranyl, pyrazinyl, pyrazolopyrimidinyl, pyrazolyl, pyridizinyl, pyridyl, pyrimidinyl, pyrrolyl, quinolinyl, tetrazolyl, thiazolyi, thienylpyrazolyl, thiophenyl, triazolyl, benzo[ ⁇ i]oxazolyl, benzofuranyl, benzothiazolyl, benzothiophenyl, be ⁇ zoxadiazolyl, dihydrobenzodioxynyl, furanyl, imidazolyl, indoiyl, and isoxazolyl.
- heterocycloalkyl is a heterocycle as described above, which is fully or partially saturated.
- preferred heterocycloalkyl groups are 5- to 7-membered heterocycloalkyi groups having a single saturated ring with 5 to 7 ring members. I or 2 ring members independently chosen from N, O and S, and remaining ring members being carbon. Certain such heterocycloalkyl groups are 5- or 6-membered.
- a "heterocyc]oalkyICo-C n aIkyl” is a heterocycloalkyi group linked via a single covalent bond or group, such as a C 1 - Cjaikylene group.
- (3- to 10-membered cycle) refers to any carbocycie or heterocycle that has from
- C ⁇ alkoxy groups include "(3- to 10-membered cycle)-O-" moieties, such as phenoxy and
- cycloalkyloxy groups e.g., cyclopentyloxy
- a "substituent,” as used herein, refers to a molecular moiety that is covalently bonded to an atom within a molecule of interest.
- a ring substituent may be a moiety such as a halogen, alkyl group, haloalkyl group or other group discussed herein that is covalently bonded to an atom (preferably a carbon or nitrogen atom) that is a ring member.
- Substituents of aromatic groups are generally covalently bonded to a ring carbon atom.
- substitution refers to replacing a hydrogen atom in a molecular structure with a substituent, such that the valence on the designated atom is not exceeded, and such that a chemically stable compound (i.e., a compound that can be isolated, characterized, and tested for biological activity) results from the substitution.
- Groups that are "optionally substituted” are unsubstituted or are substituted by other than hydrogen at one or more available positions, typically I 5 2, 3, 4 or 5 positions, by one or more suitable groups (which may be the same or different).
- Optional substitution is also indicated by the phrase "substituted with from 0 to X substituents," where X is the maximum number of possible substituents.
- Certain optionally substituted groups are substituted with from 0 to 2, 3 or 4 independently selected substituents (i.e., are unsubstituted or substituted with up to the recited maximum number of substituents).
- MCH receptor refers to any naturally-occurring mammalian (e.g., human, monkey, or canine) MCH type 1 or type 2 receptor, as well as chimeric receptors in which one or more domains of a naturally-occurring MCHlR or MCH2R are replaced with a corresponding domain of a different G protein-coupled receptor, such that the ability of the chimeric receptor to bind MCH and mediate a dose-dependent release of intracellular calcium is not diminished.
- MCH receptors for use within the various assays and other methods described herein include, for example, recombinantly expressed human MCHlR (e.g., SEQ ⁇ D NO:6 of US Patent No.
- a “MCH receptor modulator,” also referred to herein as a “modulator,” is a compound that alters (increases or decreases) MCH receptor activation and/or MCH receptor-mediated signal transduction.
- MCH receptor modulators specifically provided herein are aminopiperidines and related compounds.
- a modulator may exhibit an EC 50 or IC 5 0 at MCH receptor that is less than 1 micromolar, 500 11M, 200 nM, 100 nM, 50 nM, 25 nM or 10 nM in a standard calcium mobilization assay (as described in Example 10, herein) and/or an agonist- stimulated GTP gamma 35 S binding assay (as described in Example 8, herein).
- a modulator may be a MCH receptor agonist or antagonist, although antagonists are preferred for certain purposes described herein.
- a MCH receptor modulator binds with "high affinity” if the Kj at a MCH receptor is less than 1 micromolar, preferably less than 500 nanomolar, 100 nanomolar or 10 nanomolar.
- a modulator binds "specifically" to MCH receptor if it binds to a MCH receptor (total binding minus nonspecific binding) with a K; that is 10-fold, preferably 100-fold, and more preferably 1000-fold, less than the Kj measured for modulator binding to other G protein-coupled receptors.
- a modulator may have a K 1 of 500 nanomolar or less in an MCH receptor ligand binding assay and a
- K of at least 1 micromolar in a dopamine receptor ligand binding assay, such as the assay described in Example 8 (pages 111-1 12) of PCT International Publication Number WO 02/094799, which is hereby incorporated by reference.
- Representative assays for determining K, at MCH receptor are provided in Examples 6 and 8-10, herein.
- a modulator is considered an "antagonist" if it detectably inhibits MCH binding to MCH receptor and/or MCH-mediated signal transduction (using, for example, the representative assay provided in Example 6 or Example 9); in general, such an antagonist has a IC 50 value of less than 1 micromoiar, preferably less than 100 nanomolar, and more preferably less than 10 nanomolar within the assay provided in Example 6 and/or the assay provided in Example 9.
- MCH receptor antagonists include neutral antagonists and inverse agonists.
- An "inverse agonist” is a compound that reduces the activity of MCH receptor below its basal activity level in the absence of added ligand. Inverse agonists may also inhibit the activity of MCH at MCH receptor, and/or may also inhibit binding of MCH to MCH receptor.
- the ability of a compound to inhibit the binding of MCH to MCH receptor may be measured by a binding assay, such as the binding assays given in Example 6 or Example 9.
- the basal activity of MCH receptor, as well as the reduction in MCH receptor activity due to the presence of antagonist may be determined from a calcium mobilization assay, such as the assay of Example 10, or an agonist- stimulated GTP gamma 33 S binding assay, such as the assay described in Example 8.
- a "neutral antagonist" of MCH receptor is a compound that inhibits the activity of MCH at
- MCH receptor but does not significantly change the basal activity of the receptor (e.g., within an assay as described in Example 8 or Example 10 performed in the absence of ligand, MCH receptor activity is reduced by no more than 10%, more preferably by no more than 5%, and even more preferably by no more than 2%; most preferably, there is no detectable reduction in activity).
- Neutral antagonists may also inhibit ligand binding to MCH receptor.
- MCH receptor agonist is a compound that elevates the activity of the receptor above the basal activity level of the receptor (i.e., enhances MCH receptor activation and/or MCH receptor-mediated signal transduction).
- MCH receptor agonist activity may be identified using the representative assays provided in Examples 8 and 10. In general, such an agonist has an EC 50 value of less than 1 micromoiar, preferably less than 100 nanomolar, and more preferably less than 10 nanomolar within one or both of the assays provided in Examples 8 and 10.
- a “therapeutically effective amount” is an amount that, upon administration, is sufficient to provide a discernible patient benefit.
- a therapeutically effective amount may reduce symptom severity or frequency, and/or may result in detectable weight loss.
- a therapeutically effective amount may improve patient status or outcome and/or prevent or delay disease or symptom onset.
- a therapeutically effective amount or dose generally results in a concentration of compound in a body fluid (such as blood, plasma, serum,
- CSF synovial fluid, lymph, cellular interstitial fluid, tears or urine
- a “disease or disorder associated with MCH receptor activation,” as used herein is any condition that is characterized by inappropriate stimulation of MCH receptor, regardless of the amount of MCH present locally, and/or that is responsive to modulation of MCH receptor activity (i.e., the condition or a symptom thereof is alleviated by such modulation).
- Such conditions include, for example, metabolic disorders (such as diabetes), heart disease, stroke, eating disorders (such as obesity and bulimia nervosa) and sexual disorders such as anorgasmic and psychogenic impotence, as well as other diseases and disorders recited herein.
- a “patient” is any individual treated with an aminopiperidine or related compound as provided herein. Patients include humans, as well as other animals such as companion animals (e.g., dogs and cats) and livestock. Patients may be experiencing one or more symptoms of a condition responsive to MCH receptor modulation, or may be free of such symptom(s) (i.e., treatment may be prophylactic).
- MCH receptor modulators may be specific for a particular MCH receptor (e.g., type 1 or type 2) or may inhibit or enhance ligand binding to multiple MCH receptors.
- MCH receptor modulators may be used to modulate MCH receptor activity in vivo, especially in the treatment of metabolic, feeding and sexual disorders in humans, domesticated companion animals and livestock animals. Modulators may also be used within a variety of in vitro assays, such as assays for receptor activity, as probes for detection and localization of MCH receptors and as standards in assays of MCH binding and MCH-mediated signal transduction.
- the MCH receptor modulators provided herein detectably modulate
- MCH receptor activity at submicromoiar concentrations, preferably at subnanomolar concentrations.
- Certain aminopiperidines and related compounds further satisfy Formula II, III, IV, V, VI,
- Formula I lie Formula HId
- variables are generally as described above, f ⁇ certain embodiments of the Formulas provided herein, one or more of the variables are as follows.
- R] groups satisfy the formula -L-M, in which L and M are as noted above. It will be apparent that the "L” moiety in such groups is divalent and is directly finked both to M and to the aromatic ring that comprises U and T. If L is a single covalent bond, then the "M” moiety is linked via a single covalent bond to the aromatic ring.
- Ri is a group of the Formula Rs-O-, in which linkage is via the oxygen atom and Rg is as defined for Formula IL
- R 8 is hydrogen, Cj-Qalkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, mono- or di-(Ci-C 3 alkyl)aminoCi-C 6 alkyi or (5- to 7-membered heterocycle)C 0 -C 6 alkyl.
- R, groups are Ci-C 6 alkoxy, such as methoxy; other R
- Q is substituted carbon.
- P is CR 55 , and at least one of P and Q is substituted carbon.
- R 9 and R i5 are independently hydrogen, C r C 2 alkyl, C r C 2 alkoxy or CV Cihaloalkyl; within further embodiments, R 9 and R S 5 are independently C r C 2 aikyl, C]-C 2 alkoxy or Cj-Cihaloalkyi. Rg and Rj 5 are both methyl within other embodiments.
- the variables U and T. within certain compounds provided herein, are both CR 7 .
- each R 7 is hydrogen, C]-C 2 alkyL C r C 2 aIkoxy or Ci-C 2 haloalkyl; in further embodiments, each R 7 is hydrogen.
- R9 and Rj 5 are independently chosen from Ci-C 4 alkyl and each R 7 is independently hydrogen, C r C 2 alkyl, C r C 2 alkoxy or Q- Cjhaloalkyl.
- R 3 is hydrogen. Within other such compounds, R 3 is not H (e.g., R 3 is methyl or ethyl). Within certain such compounds, the group
- n is 0, 1 or 2.
- Representative combinations for n and m include, for example, (a) n is 0 and m is 1, 2 or 3; (b) n is 1 and m is 2 or 3; (c) n is 2 and m is 0, 1 or 2; and (d) n is 3 and m is 0 or 1 .
- Certain n and m combinations are illustrated in the following table, in which the n and m values are given along with the resulting core ring:
- Such core rings may, but need not, be substituted as provided herein (e.g., by the formation of a bridge).
- R 2 represents zero substituents.
- R? represents from 0 to 4 substituents independently chosen from Cj-dalkyl and
- two R 2 groups are taken together to form a bridge.
- variable q is 0 in certain embodiments and 1 in other embodiments.
- Representative R 10 moieties include, for example, methyl, ethyl, propyl and butyl.
- X is C(O).
- R n and R !2 are independently H, Ci-Cgalkyl, CrQalkoxy, C 2 -C ⁇ ;alky1 ether, or taken together with a R B or R 6 moiety to form a Cs-Cycycioalkyl or heterocycloalkyl. It will be apparent that, if R A is a cyclic moiety, such a cycloalkyl or heterocycloalkyl will be a ring that is fused to R A . If X is C(O)NR n , R n is preferably H, C,-C 6 alkyl or C 2 -C 6 alkyl ether.
- R A within certain aminopiperidines and related compounds of the Formulas provided herein, is (e.g., methyl, ethyl, propyl, butyl or pentyl); (e.g., trifluoroethyl); (e.g., cyclopropyl, cyc ⁇ obutyl, cyclopentyl.
- R A is (Cj- C 7 cycloalkyi)Co-C 2 alkyl within certain such compounds. Representative such compounds of
- Formula IIj Formula ITk wherein f is 0, 1 , 2 or 3.
- Representative compounds of Formula Hm further satisfy one of Formulas Hk-I to IIk-3:
- each R B is independently (i) halogen, hydroxy, nitro or cyano; or (ii) C,-C 6 alkyl, C 2 -Qalkenyl, C 2 -C 6 alkynyl, Ci-C 6 alkanoyl, C]-C 6 alkyltliio, C]-C 6 alkanoyloxy, Ci-Qalkoxycarbonyl, C
- R A is 6- to 10-membered aryl ⁇ e.g., phenyl) or 5- to 10-membered heteroaiyl (e.g., a 5- or 6-membered heteroaryl such as pyridyl, pyrimidyl, thiazolyl, thiophenyl, imidazolyl, triazolyL tetrazolyl or furanyl; or a 9- or 10-membered heteroaiyl such as indolyl, IH-indolyl, isoindolyl, benzofuranyl, dihydrobenzofuranyl.
- aryl e.g., phenyl
- 5- to 10-membered heteroaiyl e.g., a 5- or 6-membered heteroaryl such as pyridyl, pyrimidyl, thiazolyl, thiophenyl, imidazolyl, triazolyL tetrazolyl or furanyl
- benzisoxazolyl benzothiadiazolyl, benzoxadiazolyl. benzotliiophenyi, benzodioxolyl, dihydrobenzodioxinyl, quinolinyl, quinazolinyl, and the like), each of which is
- R A is H E ⁇ ⁇ D ' ; wherein A and E are independently M or CR 5 ; B, J and D are independently N or CR 6 ; each R 5 is independently: (i) hydrogen, halogen, nitro, cyano or -COOH; (ii) Cj-C 6 alkyl, C 2 -C 6 alkenyl, C 2 - C 6 alkynyl, Ci-C 6 alkanoyl, C 3 -C 6 a!kanone, C 2 -C 6 alkyl ether, C r C 6 alkoxycarbonyl, C 1 - C 6 alkylsulfonyl or Ci-C 6 haloalkyl; each of which is substituted with from 0 to 3 substituents independently chosen from hydroxy, amino, cyano, Ci-C 6 alkoxy and mono- or di-(C r C 6 alkyl)amino; or (iii) taken together with a
- each R 5 is independently: (i) hydrogen, halogen, nitro, cyano or -COOH; or (ii) CpC 6 alkyl, C 2 -C 6 alkenyl, C 2 -C ⁇ ;aikynyl, CpQalkanoyl, Cs-C ⁇ alkanone,
- C 2 -C 6 alkyl ether CpCealkoxycarbony ⁇ , CpC 6 aIkylsulfony! or CpQhaloalkyl; each of which is substituted with from O to 3 substituents independently chosen from hydroxy, amino, cyano, Cp
- each R 6 is independently: (i) hydrogen, halogen, hydroxy, nitro, cyano or-COOH; or (ii) CpQalkyl, C 2 -Qalkenyl, C 2 -Qalkynyl, CpCgalkoxy, C]- C f ialkanoyl, Cj-C ⁇ alkylthio, CpC 6 aIkanoyIoxy, Ci-C 6 alkoxycai'bonyl, CpC 6 alkyIsulfonyl, C]-
- C 2 alkyl each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CpC 4 aIkyl, C r C 4 alkoxy, CpQhaloalkyl, C r C 4 haloalkoxy, mono- or di-(C r C 6 alkyl)ammo and 5- or 6-membered heterocycloalkyi; or two adjacent R 6 groups are taken together to form a fused, 5- or 6-membered ring.
- substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CpC 4 aIkyl, C r C 4 alkoxy, CpQhaloalkyl, C r C 4 haloalkoxy, mono- or di-(C r C 6 alkyl)ammo and 5- or 6-membered heterocycloalkyi; or two adjacent R 6 groups are taken together to form a fused, 5- or 6-
- Still further compounds of Foi ⁇ nuia ⁇ lm further satisfy one of Formulas 1Im-I to l ⁇ m-3:
- At least one of B, J and D is substituted carbon.
- J is substituted carbon
- B is substituted carbon
- both J and B are substituted carbon.
- at least two, or exactly two, of B, J and D are carbon that is substituted (e.g., with a substituent independently chosen from methyl, ethyl, halogen, C]-C 2 HaIOaIkVl and C]-C 2 alkoxy).
- A, J and E are optionally substituted carbon and D is N; in certain such compounds A and E are each CH.
- R A is E-G ; E-A or D-E ; wherein A, B, E and D are independently N or CR 6 ; G is NR 6 , S or O; and R 6 is as described above.
- Representative such R A groups include, for
- R 5 and R 6 are generally as indicated above.
- each R 6 is independently: (i) hydrogen, halogen, hydroxy, nitro or cyano; or (ii) d-Qalkyl, C 2 - Qalkenyl, C 2 -C 6 alkynyl, C r C 6 alkoxy, C r C 6 alkanoyl, C r C 6 alkylthio, C r QalkanoyIoxy, C 1 - C 6 alkoxycarbony], C]-C 6 alky]sulfonyl, C,-C 6 haloaikyl, (C 3 -C 7 cycioalkyl)Co-C 2 aikyl, (4- to 7- membered heteiOcycle)C,-C 2 alkyl or phenyiCo-C 2 alkyl; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen,
- R 3 or R 6 is taken together with R n or R ]2 to form a cycloalkyl or heterocycloalkyl ring. It will be apparent that, if R A is a cyclic moiety, the cycloalkyi or heterocycloalkyl ring so formed will result in fused rings (e.g., benzodioxolyl). In further embodiments, two substituents represented by R 6 are taken together to fo ⁇ n a carbocycle or heterocycle.
- R A is 6- to 10-membered aryl or 5- to 10- membered heteroaryl, each of which is substituted with from 0 to 4 substituents independently chosen from R B .
- Representative R A groups include D . wherein A and E are independently
- N or CR 5 B, J and D are independently N or CR ⁇ ; and each R 5 and R 5 is independently: (i) hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl; (ii) C r C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 aikynyl, C,-C 6 alkoxy, C r C 6 alkanoyl, C r C 6 alkylthio, C 1 - C f ialkanoyloxy, C r C 6 alkoxycarbonyl ; CpC 6 alkylsulfonyl, CpC ⁇ haloalkyl, mono- or di-(C r C 6 alkyl)aminoC 0 -C 4 alkyl, mono- or di-(CpC 6 alkyl)aminocarbonylCo-C 4 alkyl, mono- or di-(C
- R 5 groups include (i) hydrogen, halogen, nitro, cyano or -COOH; and (ii) C,-C 6 alkyi, C r C 6 alkenyl, C 2 -C 6 alkynyl, CpQalkanoyL C 3 -C 6 aikanone, C 2 - C 6 alkyl ether, CpC 6 alkoxycarbonyl, CpCealkylsulfonyl or CpCshaloalkyl; each of which is substituted with from O to 3 substituents independently chosen from hydroxy, amino, cyano, Cj- Qaikoxy and mono- or di-(CrC 6 alky!)amino; and representative R/, groups include: (i) hydrogen, halogen, hydroxy, nitro, cyano or-COOH; and (ii) CpQalkyl, Q-Qalkenyl, Ci-C ⁇ alkynyl, C r C 6 alkoxy, C r C
- R A groups are 5-membered heteroaryl groups that are substituted with from 0 to 3 substituents independently chosen from: (i) halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl; (ii) Ci-C 6 a ⁇ kyl, CrQaSkenyl, C 2 -C 6 alkynyl, Cj-C ⁇ alkoxy, C,- Cealkanoyl, CrCgalkylthio, Cj-C ⁇ salkanoyloxy, C]-C 6 alkoxycarbonyi, Ci-Cgalkylsulfo ⁇ yl, Q- C f ehaioalkyl, mono- or di-(CrC 6 aIkyl)ammoC 0 -C 4 aIkyl, mono- or di-(Ci-C6aIky!)aminocarbonylCo- C 4 alkyl, mono- or di-(C l -C
- at least one of R 9 and R 15 is not H
- -C 2 alkyl, C]- C 2 aikoxy or C)-C 2 haloalkyl; R 9 and R, 5 are each methyl; or R 9 and R
- the variable R ⁇ in certain compounds of Formula VII, VIII or IX, is not H.
- R 8 is hydrogen, C r C 6 alky1, C 2 -C ( ,alkenyl, C 2 -C 6 alkyny], mono- or di-(C r C 8 alkyl)aminoC]-C 6 alkyl or (5- to 7-membered heterocycle)Co-C ⁇ jalkyl, each of which is substituted with from O to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosuifonyl, C 1 - C 4 alkyi, and mono- and di-(C ! -C 8 alkyl)aminoCo-C 6 a]kyl.
- Ri is methoxy.
- aminopiperidines and related compounds provided herein include, but are not limited to, those specifically described in Examples 1-4, herein. It will be apparent that the compounds recited therein are representative only, and are not intended to limit the scope of the present invention. Further, as noted above, all compounds may be present as a free base, a pharmaceutically acceptable salt (such as an acid addition salt) or other form, such as a solvate (e.g., hydrate).
- a pharmaceutically acceptable salt such as an acid addition salt
- solvate e.g., hydrate
- aminopiperidines and related compounds provided herein detectably alter (modulate) MCH binding to MCHlR and/or MCH2R, as determined using a standard in vitro MCH receptor ligand binding assay and/or functional assay. References herein to a
- MCH receptor ligand binding assay refer to either of the assays provided in Examples 6 and 9.
- the receptor is incubated with labeled MCH (or other suitable ligand) and a test compound.
- a test compound that detectably modulates binding of ligand to MCH receptor will result in a decrease or increase in the amount of label bound to the MCH receptor preparation, relative to the amount of label bound in the absence of the compound.
- such a compound will exhibit a K 1 at an MCH receptor that is less than 1 micromolar, more preferably less than 500 nM, 100 nM, 20 nM or 10 nM, within an assay performed as described in Example 6 and/or within an assay performed as described in Example 9.
- Certain preferred compounds are MCH receptor antagonists, and exhibit IC 50 values of about 4 micromolar or less, more preferably 1 micromolar or less, still more preferably about 100 nanomolar or less, or 10 nanomolar or less within a standard in vitro MCH receptor mediated calcium mobilization assay, as provided in Example 10 and/or an agonist-stimulated GTP gamma 35 S binding assay, as described in Example 8.
- aminopiperidines and related compounds provided herein may be evaluated for certain pharmacological properties including, but not limited to, oral bioavailability (preferred compounds are orally bioavailable to an extent allowing for oral doses of less than 140 mg/kg, preferably less than 50 mg/kg, more preferably less than 30 mg/kg, even more preferably less than 10 mg/kg, still more preferably less than 1 mg/kg), toxicity (a preferred compound is nontoxic when a therapeutically effective amount is administered to a subject), side effects (a preferred compound produces side effects comparable to placebo when a therapeutically effective amount of the compound is administered to a subject), serum protein binding and in vitro and in vivo half-life (a preferred compound exhibits an in vitro half-life that is equal to an in vivo half-life allowing for Q.
- oral bioavailability preferred compounds are orally bioavailable to an extent allowing for oral doses of less than 140 mg/kg, preferably less than 50 mg/kg, more preferably less than 30 mg/kg, even more preferably less than 10 mg
- LD. dosing preferably T.I.D. dosing, more preferably B. I. D. dosing, and most preferably once-a- day dosing).
- differential penetration of the blood brain barrier may be desirable for compounds used to treat CNS disorders, while low brain levels of compounds used to treat peripheral disorders are preferred.
- Routine assays that are well known in the art may be used to assess these properties and identify superior compounds for a particular use. For example, assays used to predict bioavailability include transport across human intestinal cell monolayers, including Caco ⁇ 2 cell monolayers. Penetration of the blood brain barrier of a compound in humans may be predicted from the brain levels of the compound in laboratory animals given the compound (e.g., intravenously).
- Serum protein binding may be predicted from albumin binding assays.
- Compound half-life is inversely proportional to the frequency of dosage of a compound.
- In vitro half-lives of compounds may be predicted from assays of microsomal half-life as described in Example 12.
- nontoxic shall be understood in a relative sense and is intended to refer to any substance that has been approved by the United States Food and Drug Administration (“FDA”) for administration to mammals (preferably humans) or, in keeping with established criteria, is susceptible to approval by the FDA for administration to mammals (preferably humans).
- FDA United States Food and Drug Administration
- a highly preferred nontoxic compound generally satisfies one or more of the following criteria when administered in minimum therapeutically effective amounts, or when contacted with cells at a concentration that is sufficient to inhibit the binding of Iigand to MCH receptor in vitro: (1) does not substantially inhibit cellular ATP production; (2) does not significantly prolong heart QT intervals; (3) does not cause substantial liver enlargement and (4) does not cause substantial release of liver enzymes.
- a compound that does not substantially inhibit cellular ATP production is a compound that satisfies the criteria set forth in Example 1 1. In other words, cells treated as described in Example 1 1 with 100 ⁇ M of such a compound exhibit ATP levels that are at least 50% of the ATP levels detected in untreated cells.
- such cells exhibit ATP levels that are at least 80% of the ATP levels detected in untreated cells.
- concentration of compound used in such assays is generally at least 10-fold, 100-fold or 1000-fold greater than the EC 50 or IC 50 for the modulator in the assay of Example 8 or Example 10.
- a compound that does not significantly prolong heart QT intervals is a compound that does not result in a statistically significant prolongation of heart QT intervals (as determined by electrocardiography) in guinea pigs, minipigs or dogs upon administration of a dose that yields a serum concentration equal to the ECjo or IC 50 for the compound.
- a dose of 0.01, 0.05, 0.1 , 0.5, 1, 5, 10, 40 or 50 mg/kg administered parenterally or orally does not result in a statistically significant prolongation of heart QT intervals.
- statically significant is meant results varying from control at the p ⁇ 0.1 level or more preferably at the p ⁇ 0.05 level of significance as measured using a standard parametric assay of statistical significance such as a student's T test.
- a compound does not cause substantial liver enlargement if daily treatment of laboratory rodents (e.g., mice or rats) for 5-10 days with a dose that yields a serum concentration equal to the EC 50 or IC 50 for the compound results in an increase in liver to body weight ratio that is no more than 100% over matched controls. In more highly preferred embodiments, such doses do not cause liver enlargement of more than 75% or 50% over matched controls. If non-rodent mammals (e.g., dogs) are used, such doses should not result in an increase of liver to body weight ratio of more than 50%, preferably not more than 25%, and more preferably not more than 10% over matched untreated controls. Preferred doses within such assays include 0.01, 0.05. 0.1 , 0.5, 1, 5, 10.
- a compound does not promote substantial release of liver enzymes if administration of twice the minimum dose that yields a serum concentration equal to the EC J O or IC 50 for the compound does not elevate serum levels of ALT, LDH or AST in laboratory rodents by more than 100% over matched mock-treated controls. In more preferred embodiments, such doses do not elevate such serum levels by more than 75% or 50% over matched controls.
- a compound does not promote substantial release of liver enzymes if, in an in vitro hepatocyte assay, concentrations (in culture media or other such solutions that are contacted and incubated with hepatocytes in vitro) that are equal to the EC 50 or IC 50 for the compound do not cause detectable release of any of such liver enzymes into culture medium above baseline levels seen in media from matched mock-treated control cells. In more highly preferred embodiments, there is no detectable release of any of such liver enzymes into culture medium above baseline levels when such compound concentrations are five-fold, and preferably ten-fold, the EC 50 or IC 5 Q for the compound.
- certain preferred compounds do not inhibit or induce microsomal cytochrome P450 enzyme activities, such as CYP 1A2 activity, CYP2A6 activity.
- CYP2C9 activity, CYP2C19 activity, CYP2D6 activity, CYP2E1 activity or CYP3A4 activity at a concentration equal to the EC50 or IC50 for the compound.
- Certain preferred compounds are not clastogenic (e.g., as determined using a mouse erythrocyte precursor cell micronucieus assay, an Ames micronucleus assay, a spiral micronucleus assay or the like) at a concentration equal the EC 50 or IC 50 for the compound.
- certain preferred compounds do not induce sister chromatid exchange (e.g., in Chinese hamster ovaiy cells) at such concentrations.
- aminopiperi dines and related compounds provided herein may be isotopically-iabeled or radiolabeled.
- compounds of Formula I may have one or more atoms replaced by an atom of the same element having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be present in the compounds provided herein include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2 H, ⁇ , 1 1 C, ' J C, 1 4 C. 15 N, ' 8 O, 17 O, 31 P, 32 P, 35 S, 18 F and 36 Cl.
- substitution with heavy isotopes such as deuterium (i.e., " H) can afford certain therapeutic advantages resulting from greater metabolic stability, such as increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some circumstances.
- Aminopiperidines and related compounds can be administered as the neat chemical, but are preferably administered as a pharmaceutical composition comprising such a compound, together with at least one physiologically acceptable carrier or excipient.
- Representative carriers include, for example, water, buffers (e.g., neutral buffered saline or phosphate buffered saline), ethanof, mineral oil, vegetable oil, dimethyl sulfoxide, carbohydrates (e.g., glucose, mannose, sucrose or dextrans), mannitol and proteins.
- Additional optional components include adjuvants, diluents, polypeptides or amino acids such as glycine, antioxidants, chelating agents such as EDTA or glutathione and/or preservatives.
- Preferred pharmaceutical compositions are formulated for oral delivery to humans or other animals (e.g., companion animals such as dogs or cats).
- other active ingredients may (but need not) be included in the pharmaceutical compositions provided herein.
- Pharmaceutical compositions may also optionally comprise an activity enhancer, chosen from a wide variety of molecules that function in different ways to enhance MCH receptor modulator effect. Particular classes of activity enhancers include skin penetration enhancers and absorption enhancers, Pharmaceutical carriers must be of sufficiently high purity and sufficiently low toxicity to render them suitable for administration to the animal being treated.
- the carrier can be inert or it can possess pharmaceutical benefits.
- the amount of carrier employed in conjunction with the compound is sufficient Io provide a practical quantity of material for administration per unit dose of the compound.
- Representative pharmaceutically acceptable carriers or components thereof are sugars, such as lactose, giucose and sucrose; starches, such as corn starch and potato starch; cellulose and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and methyl cellulose; powdered tragacanth; malt; gelatin: talc; solid lubricants, such as stearic acid and magnesium stearate; calcium sulfate; synthetic oils; vegetable oils, such as peanut oil, cottonseed oil, sesame oil, olive oil and corn oil; polyols such as propylene glycol, glycerine, sorbitol, mannitol and polyethylene glycol; alginic acid; phosphate buffer solutions; emuisifiers, such as the TWEENS; we
- a pharmaceutical composition effective concentrations of one or more aminopiperidines or related compounds provided herein are mixed with one or more suitable pharmaceutical earners or excipients.
- methods for solubilizing compounds may be used. Such methods are known to those of skill in this art and include, but are not limited to, using cosolvents such as dimethylsulfoxide (DMSO), using a surfactant, such as TWEEN, or dissolution in aqueous sodium bicarbonate.
- DMSO dimethylsulfoxide
- TWEEN a surfactant
- dissolution in aqueous sodium bicarbonate dissolution in aqueous sodium bicarbonate.
- the resulting mixture may be a solution, suspension, emulsion or the like. The form of the resulting mixture depends upon a number of factors, including the intended mode of administration and the solubility of the compound in the chosen carrier.
- compositions may be formulated for administration by any suitable route, including orally, topically, parenterally, by inhalation (e.g., nasal or oral) or spray, sublingually, transdermally, via buccal administration, rectal Iy, as an ophthalmic solution or by other means, and may be prepared in dosage unit formulations and/or formulated as a lyophilizate.
- parenteral as used herein includes subcutaneous, intradermal, intravascular (e.g., intravenous), intramuscular, spinal, intracranial, intrathecal and intraperitoneal injection, as well as any similar injection or infusion technique.
- Dosage formulations suitable for oral use include, for example, tablets, troches, lozenges, liquid solutions, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, tinctures, syrups or elixirs.
- Typical components of carriers for syrups, elixirs, emulsions and suspensions include ethanol, glycerol, propylene glycol, polyethylene glycol, liquid sucrose, sorbitol and water. Such formulations may also contain a demulcent.
- formulations containing these compounds can be presented as a dry product (optionally as an admixture with a dispersing or wetting agent, suspending agent and one or more preservatives) for constitution with water or other suitable vehicle before use.
- Aqueous suspensions comprise the active materials ) in admixture with one or more suitable excipients, such as suspending agents (e.g., sodium carboxymethylcellulose, methylcellulose, hydroxypropyjmethylcellulose, AVICEL RC-591, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia); and dispersing or wetting agents (e.g., polysorbate 80, naturaily- occurring phosphatides such as lecithin, condensation products of an alkylene oxide with fatty acids such as polyoxyethylene stearate, condensation products of ethylene oxide with long chain aliphatic alcohols such as heptadecaethyleneoxycetanol, condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate. or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides such as polyethylene sorbitan monoo
- Oily suspensions may be formulated by suspending the active ingredients in a vegetable oil (e.g., peanut oil, olive oil, sesame oil or coconut oil), a mineral oil (such as liquid paraffin) or a mixture of such oils.
- the oily suspensions may further contain a thickening agent, such as beeswax, hard paraffin or cetyl alcohol.
- compositions provided herein may also be in the form of oil-in-water emulsions.
- the oily phase may be a vegetable oil, mineral oil, or mixture thereof as described above.
- Suitable emulsifying agents include naturally-occurring gums (e.g., gum acacia or gum tragacanth), naturally-occurring phosphatides (e.g., soy bean phosphatide, lecithin and esters or partial esters derived from fatty acids and hexitol), and anhydrides (e.g., sorbitan monoleate and condensation products of the above partial esters with ethylene oxide, such as polyoxyethylene sorbitan monoleate).
- naturally-occurring gums e.g., gum acacia or gum tragacanth
- naturally-occurring phosphatides e.g., soy bean phosphatide, lecithin and esters or partial esters derived from fatty acids and hexitol
- anhydrides e
- Excipients suitable for the manufacture of tablets and capsules include, for example, inert diluents to increase the bulk weight of the material to be tableted (e.g., calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate), granulating and disintegrating agents that modify the disintegration rate in the environment of use (e.g., corn starch, starch derivatives, alginic acid and salts of carboxymethylcellulose), binding agents that impart cohesive qualities to the powdered material(s) (e.g., starch, gelatin, acacia and sugars such as sucrose, glucose, dextrose and lactose) and lubricating agents (e.g., magnesium stearate, calcium stearate, stearic acid or talc).
- inert diluents to increase the bulk weight of the material to be tableted e.g., calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate
- GIidants such as silicon dioxide can be used to improve flow characteristics of the powder mixture.
- Tablets may be formed using standard techniques, including dry granulation, direct compression and wet granulation. Tablets may be uncoated or may be coated by known techniques.
- Capsules include, for example, hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent (e.g., calcium carbonate, calcium phosphate or kaolin), as well as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium (e.g., peanut oil, liquid paraffin or olive oil).
- an inert solid diluent e.g., calcium carbonate, calcium phosphate or kaolin
- an oil medium e.g., peanut oil, liquid paraffin or olive oil
- Compositions intended for oral use may further contain one or more optional agents, such as sweetening agents (e.g., glycerol, propylene glycol, sorbitol, sucrose, aspartame, saccharin, menthol, peppermint or fruit flavor); suspending agents (e.g., sorbitol syrup, methyl cellulose, glucose/sugar, syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminum stearate gel and hydrogenated edible fats); emulsifying agents (e.g., lecithin, sorbitan monsoleate or acacia); nonaqueous vehicles such as edible oils (e.g., almond oil, fractionated coconut oil, silyl esters, propylene glycol and ethyl alcohol); preservatives (e.g., methyl or propyl p-hydroxybenzoate, sodium benzoate, methyl paraben, ascorbic acid and/or sorbic acid); flavoring agents; and/
- a pharmaceutical composition may be prepared as a sterile injectable aqueous or oleaginous suspension.
- the active ingredient(s), depending on the vehicle and concentration used, can either be suspended or dissolved in the vehicle.
- Such a composition may be formulated according to the known art using suitable dispersing, wetting agents and/or suspending agents such as those mentioned above.
- suitable dispersing, wetting agents and/or suspending agents such as those mentioned above.
- suitable dispersing, wetting agents and/or suspending agents such as those mentioned above.
- suitable dispersing, wetting agents and/or suspending agents such as those mentioned above.
- the acceptable vehicles and solvents that may be employed are water,
- 1,3-butanedioI Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils may be employed as a solvent or suspending medium.
- any bland fixed oil may be employed, including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectable compositions, and adjuvants such as local anesthetics, preservatives and/or buffering agents can be dissolved in the vehicle.
- compositions may also be prepared in the form of suppositories (e.g., for rectal administration).
- Such compositions can be prepared by mixing the drug with a suitable non- irritating excipient that is solid at ordinary temperatures but liquid at the body temperature and will therefore melt in the body to release the drug.
- suitable excipients include, for example, cocoa butter and polyethylene glycols.
- compositions for inhalation typically can be provided in the form of a solution, suspension or emulsion that can be administered as a dry powder or in the form of an aerosol using a conventional propel! ant (e.g., dichlorodifiuoromethane or trichiorofluoromethane).
- a conventional propel! ant e.g., dichlorodifiuoromethane or trichiorofluoromethane.
- compositions may be formulated for local or topical application, such as for topical application to the skin or mucous membranes.
- Topical compositions may be in any suitable form including, for example, solutions, creams, ointments, gels, lotions, milks, cleansers, moisturizers, sprays, skin patches and the like. Such solutions may, for example, be formulated as 0.01 %- 10% isotonic solutions, pH about 5-7, with appropriate salts.
- Pharmaceutical compositions may also be formulated for transdermal administration as a transdermal patch. Pharmaceutical compositions may be formulated for release at a pre-determined rate.
- Instantaneous release may be achieved, for example, via sublingual administration (i.e., administration by mouth in such a way that the active ingredient(s) are rapidly absorbed via the blood vessels under the tongue rather than via the digestive tract).
- Controlled release formulations i.e., formulations such as a capsule, tablet or coated tablet that slows and/or delays release of active ingredient(s) following administration
- a controlled release formulation comprises a matrix and/or coating that delays disintegration and absorption in the gastrointestinal tract (or implantation site) and thereby provides a delayed action or a sustained action over a longer period.
- One type of controlled-release formulation is a sustained-release formulation, in which at least one active ingredient is continuously released over a period of time at a constant rate.
- the therapeutic agent is released at such a rate that blood (e.g., plasma) concentrations are maintained within the therapeutic range, but below toxic levels, over a period of time that is at least 4 hours, preferably at least 8 hours, and more preferably at least 12 hours.
- Such formulations may generally be prepared using well known technology and administered by, for example, oral, rectal or subcutaneous implantation, or by implantation at the desired target site.
- Carriers for use within such formulations are biocompatible, and may also be biodegradable; preferably the formulation provides a relatively constant level of modulator release.
- the amount of active ingredient contained within a sustained release formulation depends upon, for example, the site of implantation, the rate and expected duration of release and the nature of the condition to be treated or prevented.
- Controlled release may be achieved by combining the active ingredient(s) with a matrix material that itself alters release rate and/or through the use of a controiled-release coating.
- the release rate can be varied using methods well known in the art, including (a) varying the thickness or composition of coating, (b) altering the amount or manner of addition of plasticizer in a coating, (c) including additional ingredients, such as release-modifying agents, (d) altering the composition, particle size or particle shape of the matrix, and (e) providing one or more passageways through the coating.
- the amount of modulator contained within a sustained release formulation depends upon, for example, the method of administration (e.g., the site of implantation), the rate and expected duration of release and the nature of the condition to be treated or prevented.
- the matrix material which itself may or may not serve a controlled-release function, is generally any material that supports the active ingredient(s).
- a time delay material such as glyceryl monosterate or glyceryl distearate may be employed.
- Active ingredient(s) may be combined with matrix material prior to formation of the dosage form (e.g., a tablet).
- active ingredient(s) may be coated on the surface of a particle, granule, sphere, microsphere, bead or pellet that comprises the matrix material. Such coating may be achieved by conventional means, such as by dissolving the active ingredient(s) in water or other suitable solvent and spraying.
- additional ingredients are added prior to coating (e.g., to assist binding of the active ingredient(s) to the matrix material or to color the solution).
- the matrix may then be coated with a barrier agent prior to application of controlled-release coating. Multiple coated matrix units may, if desired, be encapsulated to generate the final dosage form.
- a controlled release is achieved through the use of a controlled release coating (i.e., a coating that permits release of active ingredient(s) at a controlled rate in aqueous medium).
- the controlled release coating should be a strong, continuous film that is smooth, capable of supporting pigments and other additives, non-toxic, inert and tack-free. Coatings that regulate release of the modulator include pH-independent coatings, pH-dependent coatings
- enteric coatings which allow the formulation to pass intact through the stomach and into the small intestine, where the coating dissolves and the contents are absorbed by the body.
- multiple coatings may be employed (e.g., to allow release of a portion of the dose in the stomach and a portion further along the gastrointestinal tract).
- a portion of active ingredient(s) may be coated over an enteric coating, and thereby released in the stomach, while the remainder of active ingredient(s) in the matrix core is protected by the enteric coating and released further down the GI tract.
- pH dependent coatings include, for example, shellac, cellulose acetate phthalate, polyvinyl acetate phthalate, hydroxypropylmethylcellulose phthalate. methacrylic acid ester copolymers and zein.
- the coating is a hydrophobic material, preferably used in an amount effective to slow the hydration of the gelling agent following administration.
- Suitable hydrophobic materials include alkyi celluloses (e.g., ethylcellulose or carboxymethylcellulose), cellulose ethers, cellulose esters, acrylic polymers (e.g., poly( acrylic acid), poly(methacrylic acid), acrylic acid and methacrylic acid copolymers, methyl methacrylate copolymers, ethoxy ethyl meth aery 1 ate s, cyanoethyl methacrylate.
- alkyi celluloses e.g., ethylcellulose or carboxymethylcellulose
- cellulose ethers e.g., cellulose ethers, cellulose esters
- acrylic polymers e.g., poly( acrylic acid), poly(methacrylic acid), acrylic acid and methacrylic acid copolymers, methyl methacrylate copolymers, ethoxy
- aqueous dispersions of ethylceliulose include, for example,
- AQUACOAT® FMC Corp., Philadelphia, PA
- SURELEASE® Colorcon, Inc., West Point
- PA both of which can be applied to the substrate according to the manufacturer's instructions.
- Acrylic polymers include, for example, the various EUDRAGIT® (Rohm America,
- Piscataway, NJ Polymers, which may be used singly or in combination depending on the desired release profile, according to the manufacturer's instructions.
- Suitable plasticizers for alkyi celluloses include, for example, dibutyl sebacate, diethyl phthalate, triethyl citrate, tributyl citrate and triacetin.
- Suitable piasticizers for acrylic polymers include, for example, citric acid esters such as triethyl citrate and tributyl citrate, dibutyl phthalate, polyethylene glycols, propylene glycol, diethyl phthalate, castor oil and triacetin.
- Controiled-release coatings are generally applied using conventional techniques, such as by spraying in the form of an aqueous dispersion, if desired, the coating may comprise pores or channels or to facilitate release of active ingredient. Pores and channels may be generated by well known methods, including the addition of organic or inorganic material that is dissolved, extracted or leached from the coating in the environment of use.
- pore-forming materials include hydrophilic polymers, such as hydroxyalkylcelluloses (e.g., hydroxypropylmethylcellulose), cellulose ethers, synthetic water-soluble polymers (e.g., polyvinylpyrrolidone, cross-linked polyvinylpyrrolidone and polyethylene oxide), water-soluble polydextrose, saccharides and polysaccharides and alkali metal salts.
- a controlled release coating may include one or more orifices, which may be formed my methods such as those described in US Patent Nos. 3,845,770; 4,034,758; 4,077,407; 4,088,864; 4,783,337 and 5,071,607. Controiled- release may also be achieved through the use of transdermal patches, using conventional technology (see, e.g., US Patent No. 4,668,232).
- controlled release formulations and components thereof, may be found, for example, in US Patent Nos. 4,572,833; 4,587,117; 4,606,909; 4,610,870; 4,684,516; 4,777,049;
- an aminopiperidine or related compound may be conveniently added to food or drinking water, for administration to humans or non-human animals including companion animals, such as dogs and cats, and livestock.
- Animal feed and drinking water compositions may be formulated so that the animal takes in an appropriate quantity of the composition along with its diet. It may also be convenient to present the composition as a premix for addition to feed or drinking water.
- Aminopiperidines and related compounds are generally present within a pharmaceutical composition at levels providing a therapeutically effective amount upon administration, as described above.
- Dosage forms providing dosage levels ranging from about 0.1 mg to about 140 mg per kilogram of body weight per day are preferred (about 0.5 mg to about 7 g per human patient per day), with dosages ranging from 0.1 mg to 50 mg, 30 mg or 10 mg particularly preferred.
- the amount of active ingredient that may be combined with the carrier to produce a single dosage form will vary depending upon the patient to be treated and the particular mode of administration.
- Dosage unit forms generally contain from about 1 mg to about 500 mg of an active ingredient.
- Dosage units generally contain from about 10 ⁇ g to about 500 mg of each active ingredient. Optimal dosages may be established using routine testing and procedures that are well known in the art.
- compositions provided herein may also contain additional active agents, which can be chosen from a wide variety of molecules and can function in different ways to enhance the therapeutic effects of a MCH receptor modulator, or to provide a separate therapeutic effect that does not substantially interfere with the activity of the MCH receptor modulator.
- additional active agents when present, are typically employed in the compositions described herein at a level ranging from about 0.01% to about 50% by weight of the composition, preferably 0.1% to 25%, 0.2% to 15, 0.5% to 10% or 0.5% to 5% by weight of the composition.
- compositions intended for the treatment of obesity and/or eating disorders may further comprise leptin, a leptin receptor agonist, a melanocortin receptor 4 (MC4) agonist, sibutramine, dexfenfluramine, a growth hormone secretagogue, a beta-3 agonist, a 5HT-2 agonist, an orexin antagonist, a neuropeptide Yi or Y 5 antagonist, a galanin antagonist, a CCK agonist, a GLP-I agonist, a cannabinoid receptor antagonist ⁇ e.g., a CB 3 antagonist) and/or a corticotropin-releasing hormone agonist.
- leptin a leptin receptor agonist
- a melanocortin receptor 4 (MC4) agonist sibutramine
- dexfenfluramine a growth hormone secretagogue
- beta-3 agonist a beta-3 agonist
- 5HT-2 agonist an orexin antagonist
- a neuropeptide Yi or Y 5 antagonist
- an additional active agent is a CBl antagonist.
- Representative CBl antagonists include, for example, certain pyrimidines ⁇ e.g., PCT International Application Publication No. WO 04/029,204), pyrazines ⁇ e.g., PCT International Application Publication Nos. WO 01 /1 1 1 ,038; WO 04/1 1 1,034 and WO 04/11 1 ,033). azetidine derivatives ⁇ e.g., US Patent Nos.
- WO 03/063781 and WO 03/040107 substituted furo[2,3-b]pyridine derivatives ⁇ e.g., PCT International Application Publication No. WO 04/012671); substituted aryl amides ⁇ e.g., PCT International Application Publication Nos. WO 03/087037 and WO 03/077847); substituted bicyclic or spirocyclic amides ⁇ e.g., PCT International Application Publication Nos. WO 03/086288 and WO 03/082190); and substituted 2,3-diphenyl pyridines ⁇ e.g , PCT International Application Publication No. WO 03/08219 ] ).
- CBl antagonists are cannabidiol and its derivatives.
- Preferred CB l antagonists include, for example, aryl substituted pyrazole carboxamides such as SR-141716A (N- ⁇ iperidin-1- yl)-5-(4-chIoropheny1)-l-(2,4-dichloro ⁇ henyl)-4-methyi- 1 -H-pyrazole-3-carboxamide) as well analogues thereof such as AM251 (N-piperidin-l-yl)-5-(4-iodophenyl)-l-(2,4-dichlorophenyl)-4- methyl- 1 -H-pyrazole-3-carboxamide) and AM281 (N-(morphoIin-4-yl)-] -(2,4-dichioiOphenyl)-5-(4- iodophenyl)-4-methyl-l -H-pyrazole-3-carboxamide); various
- compositions may be packaged for treating or preventing a disease or disorder that is associated with MC ⁇ receptor activation (e.g., treatment of metabolic disorders such as diabetes, heart disease, stroke, metabolic syndrome, obesity and eating disorders such as bulimia, skin disorders such as vitiligo, or sexual disorders such as anorgasmic or psychogenic impotence), or for promoting weight loss.
- Packaged pharmaceutical preparations comprise a container holding a therapeutically effective amount of MC ⁇ receptor modulator as described herein and instructions (e.g., labeling) indicating that the contained composition is to be used for promoting weight loss or for treating or preventing a disease or disorder that is associated with MC ⁇ receptor activation in the patient.
- Prescribing information may be provided separately to a patient or health care provider, or may be provided as a label or package insert. Prescribing information may include, for example, efficacy, dosage and administration, contraindication and adverse reaction information pertaining to the pharmaceutical formulation. Certain packaged pharmaceutical preparations further include a second therapeutic agent, as discussed above.
- the present invention provides methods for treating, preventing, or inhibiting the development or progression of a disease or disorder responsive to MCH receptor modulation.
- therapeutic methods provided herein may be used to treat a patient already afflicted with such a disease or disorder, or may be used to prevent or delay the onset of such a disease or disorder in a patient who is free of detectable disease or disorder that is associated with MCH receptor activation.
- a disease or disorder is "associated with MCH receptor activation " ' if it is characterized by inappropriate stimulation of MCH receptor, regardless of the amount of MCH present locally, and/or is responsive to modulation of MCH receptor activity.
- Such conditions include, for example, metabolic disorders (such as diabetes), metabolic syndrome, heart disease, stroke, eating disorders (such as obesity and bulimia nervosa), disorders of the skin such as vitiligo, and sexual disorders such as anorgasmic or psychogenic impotence. These conditions may be diagnosed and monitored using criteria that have been established in the art.
- MCH antagonists provided herein may be used to promote weight loss in patients
- MCH agonists provided herein may be used to promote weight gain in patients.
- Patients may include humans, domesticated companion animals (pets, such as dogs and cats) and livestock animals, with dosages and treatment regimes as described above.
- MCH receptor activation includes cognitive impairment and memory disorders, such as Alzheimer's disease, Parkinson's disease. mild cognitive impairment (MCI), age-related cognitive decline (ARCD), stroke, traumatic brain injury, AIDS associated dementia, and dementia associated with depression, anxiety and psychosis (including schizophrenia and hallucinatory disorders);
- MCI mild cognitive impairment
- ARCD age-related cognitive decline
- stroke traumatic brain injury
- AIDS associated dementia dementia associated with depression
- anxiety and psychosis including schizophrenia and hallucinatory disorders
- Anxiety, depression and other mood disorders including general anxiety disorder (GAD), agoraphobia, panic disorder with and without agoraphobia, social phobia, specific phobia, post traumatic stress disorder, obsessive compulsive disorder (OCD), dysthymia, adjustment disorders with disturbance of mood and anxiety, separation anxiety disorder, anticipatory anxiety acute stress disorder, adjustment disorders and cyclothymia; Reward system disorders such as addiction (e.g., opioid, nicotine or alcohol); Pain such as migraine, peripheral inflammatory pain, neuropathic pain and sympathetic nervous system associated pain; and
- Peripheral indications such as respiratory disorders (e.g., asthma), urinary disorders (e.g., urinary incontinence), gastrointestinal disorders, reproductive function disorders and cardiovascular disorders (e.g., arteriosclerosis and hypertension).
- respiratory disorders e.g., asthma
- urinary disorders e.g., urinary incontinence
- gastrointestinal disorders e.g., reproductive function disorders
- cardiovascular disorders e.g., arteriosclerosis and hypertension
- Frequency of dosage may vary depending on the compound used and the particular disease to be treated or prevented. In general, for treatment of most disorders, a dosage regimen of 4 times daily or less is preferred. For the treatment of eating disorders and obesity, a dosage regimen of 1 or
- methods for treating a patient comprising diagnosing the patient as having a disease or disorder associated with MCH receptor activation, correlating the diagnosis of the disease or disorder with the need for MCH modulator administration, and administering an a effective amount of an aminopiperidine or related compound provided herein.
- a method for treating a patient comprising administering an effective amount of an aminopiperidine or related compound to a patient having a disease or disorder associated with MCH receptor activation is also provided herein.
- the disease or disorder associated with MCH receptor activation is obesity, metabolic syndrome, an eating disorder, a sexual disorder, diabetes, heart disease or stroke.
- aminopiperidine or related compound is administered orally, intranasally, intravenously or topically.
- MCH receptor modulators provided herein may be used within combination therapy for the treatment of conditions associated with MCH receptor modulation.
- a MCH receptor modulator is administered to a patient along with a second therapeutic agent that is not primarily a MCH receptor modulator, but that is appropriate for treatment of the condition(s) of interest.
- the MCH receptor modulator and second therapeutic agent(s) may be present in the same pharmaceutical composition, or may be administered separately in either order. Suitable second therapeutic agents include those listed above.
- Suitable dosages for MCH receptor modulator(s) within such combination therapy are generally as described herein. Dosages and methods of administration of other therapeutic agents can be found, for example, in the manufacturer's instructions in the Physician's Desk Reference.
- the combination administration results in a reduction of the dosage of the second therapeutic agent required to produce a therapeutic effect ⁇ i.e., a decrease in the minimum therapeutically effective amount).
- the dosage of second therapeutic agent in a combination or combination treatment method of the invention is less than the maximum dose advised by the manufacturer for administration of the second therapeutic agent without combination administration of a MCH receptor modulator.
- this dosage is less than 3 Zi, even more preferably less than 14, and highly preferably, less than VA of the maximum dose, while most preferably the dose is less than 10% of the maximum dose advised by the manufacturer for administration of the second therapeutic agent(s) when administered without combination administration of a MCH receptor modulator. It will be apparent that the dosage amount of MCH receptor modulator component of the combination needed to achieve the desired effect may similarly be affected by the dosage amount and potency of the second therapeutic agent component of the combination.
- the combination administration of a MCH receptor modulator with a second therapeutic agent is accomplished by packaging one or more MCH receptor modulators and one or more second therapeutic agents in the same package, either in separate containers within the package or in the same container as a mixture of one or more MCH receptor modulators and one or more second therapeutic agents.
- Preferred mixtures are formulated for oral administration ⁇ e.g., as pills, capsules, tablets or the like).
- the package comprises a label or package insert indicating that the one or more MCH receptor modulators and one or more second therapeutic agents are to be taken together for the treatment of a condition that is associated with MCH receptor activation, such as obesity.
- one or more MCH receptor modulators provided herein are used along with one or more CBl antagonists within a combination therapy. Such combinations are of particular use for weight management, to reduce appetite and/or food intake or to prevent or treat obesity (e.g., promote weight loss).
- Patients may include humans, domesticated companion animals and livestock animals, with dosages and treatment regimes as described above.
- the MCH receptor modulator(s) may be administered to the patient at the same time as the CBl antagonist(s) (e.g., as a single dosage unit), or may be administered separately (before or after CBl antagonist).
- the MCH receptor modulators) and CBl antagonist s) are ultimately simultaneously present at effective concentrations in a body fluid (e.g., blood) of the patient.
- An effective concentration of MCH receptor modulator or CBl antagonist is a concentration that is sufficient to reduce one or more of food consumption, appetite and/or body mass index in the patient when repeatedly coadministered as described herein.
- the present invention provides a variety of in vitro uses for the compounds provided herein.
- such compounds may be used as probes for the detection and localization of MCH receptors, in samples such as tissue sections, as positive controls in assays for receptor activity, as standards and reagents for determining the ability of a candidate agent to bind to MCH receptor, or as radiotracers for positron emission tomography (PET) imaging or for single photon emission computerized tomography (SPECT).
- PET positron emission tomography
- SPECT single photon emission computerized tomography
- Such assays can be used to characterize MCH receptors in living subjects.
- Compounds provided herein are also useful as standards and reagents in determining the ability of a test compound to bind to MCH receptor.
- a sample may be incubated with a compound as provided herein under conditions that permit binding of the compound to MCH receptor.
- the amount of compound bound to MCH receptor in the sample is then detected.
- a compound may be labeled using any of a variety of well-known techniques (e.g., radiolabeled with a radionucleide such as tritium, as described herein), and incubated with the sample (which may be. for example, a preparation of cultured cells, a tissue preparation or a fraction thereof).
- a suitable incubation time may generally be determined by assaying the level of binding that occurs over a period of time.
- unbound compound is removed, and bound compound detected using any method for the label employed (e.g., autoradiography or scintillation counting for radiolabeled compounds; spectroscopic methods may be used to detect luminescent groups and fluorescent groups).
- a matched sample may be simultaneously contacted with radiolabeled compound and a greater amount of unlabeled compound. Unbound labeled and unlabeled compound is then removed in the same fashion, and bound label is detected. A greater amount of detectable label in the test sample than in the control indicates the presence of MCH receptor in the sample.
- Detection assays including receptor autoradiography (receptor mapping) of MCH receptors in cultured cells or tissue samples may be performed as described by Kuhar in sections 8.1.1 to 8.1.9 of Current Protocols In Pharmacology (1998) John Wiley & Sons, New York.
- Compounds provided herein may also be used within a variety of well-known cell culture and cell separation methods.
- compounds may be linked to the interior surface of a tissue culture plate or other cell culture support, for use in immobilizing MCH receptor-expressing cells for screens, assays and growth in culture.
- Compounds may also be used to facilitate ceil identification and sorting in vitro, permitting the selection of cells expressing a MCH receptor.
- the compound(s) for use in such methods are labeled as described herein.
- a compound linked to a fluorescent marker such as fluorescein
- FACS fluorescence activated cell sorting
- methods for modulating binding of MCH to an MCH receptor in vitro or in vivo, comprising contacting a MCH receptor with a sufficient amount of a modulator provided herein, under conditions suitable for binding of MCH to the receptor.
- MCH binding to receptor is inhibited by the modulator.
- the MCH receptor may be present in solution, in a cultured or isolated cell preparation or within a patient.
- the MCH receptor is a MCHlR receptor present in the hypothalamus.
- the amount of compound contacted with the receptor should be sufficient to modulate MCH binding to MCH receptor in vitro within, for example, a binding assay as described in Example 6 and/or Example 9.
- MCH receptor preparations used to determine in vitro binding may be obtained from a variety of sources, such as from HEK 293 cells or Chinese Hamster Ovary (CHO) cells transfected with a MCH receptor expression vector, as described herein.
- the MCH receptor may be present in solution, in a cultured or isolated cell preparation or within a patient.
- the amount of modulator contacted with the receptor should be sufficient to modulate MCH receptor signal transducing activity in vitro within, for example, a calcium mobilization assay as described in Example 30 and/or an agonist-stimulated GTP gamma 35 S binding assay as described in Example 8.
- An effect on signal-transducing activity may be assessed as an alteration in the electrophy ⁇ iology of the cells, using standard techniques, such as intracellular patch clamp recording or patch clamp recording. If the receptor is present in an animal, an alteration in the electrophysiology of the cell may be detected as a change in the animal's feeding behavior.
- Aminopiperidines and related analogues provided herein may generally be prepared using standard synthetic methods. Starting materials are generally readily available from commercial sources, such as Sigma-Aldrich Corp. (St. Louis, MO). For example, a synthetic route similar to that shown in any one of the following Schemes may be used. It will be apparent that the finai product and any intermediate(s) shown in the following schemes may be extracted, dried, filtered and/or concentrated, and may be further purified (e.g., by chromatography). Each variable (e.g , "R”) in the following Schemes, refers to any group consistent with the description of the compounds provided herein.
- compounds provided herein may contain one or more asymmetric carbon atoms, so that the compounds can exist in different stereoisomeric forms.
- These compounds can be, for example, racemates or optically active forms.
- ail stereoisomers are encompassed by the present invention. Nonetheless, it may be desirable to obtain single enantiomers (Le., optically active forms).
- Standard methods for preparing single enantiomers include asymmetric synthesis and resolution of the racemates. Resolution of the racemates can be accomplished, for example, by conventional methods such as crystallization in the presence of a resolving agent, or chromatography using, for example, a chiral HPLC column.
- R3 is methyl
- Each isotope is preferably carbon (e.g., 14 C), hydrogen (e.g., 3 H or
- Tritium labeled compounds may also be prepared catalytically via platinum-catalyzed exchange in tritiated acetic acid, acid-catalyzed exchange in tritiated trifluoroacetic acid, or exchange with tritium gas under heterogeneous catalysis using the compound as substrate.
- certain precursors may be subjected to tritium- halogen exchange with tritium gas, tritium gas reduction of unsaturated bonds, or reduction using sodium borotritide, as appropriate.
- Preparation of radiolabeled compounds may be conveniently performed by a radioisotope supplier specializing in custom synthesis of radiolabeled probe compounds.
- HPLC and MS conditions are as follows. Unless otherwise specified, mass spectroscopy data in the following Examples is obtained via HPLC MS Method 1. Where a different method is used, the method is indicated. Method 1
- Sample volume of 1 microliter is injected onto a 50x4.6mm Chromolith SpeedROD RP-I Se column (Merck KGaA, Darmstadt, Germany), and eluted using a 2- ⁇ hase linear gradient at a flow rate of 6 ml/min. Sample is detected using total absorbance count over the 220-340nm UV range.
- the elution conditions are: Mobile Phase A - 95% water, 5% MeOH with 0.05% TFA; Mobile Phase B - 5% water, 95% MeOH with 0.025% TFA.
- the following gradient is used: 0-0.5 min 10- 100%B, hold at ⁇ 00%B to 1.2 min, return to 10%B at 1.2 ] min. Inject to inject cycle is 2.15 min.
- Method 3 The conditions and procedure are the same as for Method 1, except that the following gradient is used: 0-0.5 min 5-300%B, hold at 100%B to 1.2 min, return to 5%B at 1.21 min. Inject to inject cycle is 2.15 min. Method 3
- Sample volume of 1-10 microliter is injected onto a 30x4.6mm XBridge Cl 8 5 micron column or equivalent, and eluted using a 2-phase linear gradient at a flow rate of 4 ml/min. Sample is detected using total absorbance count over the 220-254 UV range.
- the elution conditions are: Mobile Phase A - 95% water, 5% MeOH with 0.1% ammonium hydroxide; Mobile Phase B - 5% water, 95% MeOH with 0.025% formic acid. The following gradient is used: 0.01-2.00 min 0- 100% B, hold at 100%B to 3.50 min, return to 0%B at 3.51 min.
- This Example illustrates the preparation of certain representative aminopiperidines and related compounds.
- Step 1 8-[4-(2-Methoxy-ethoxy)-2,3-dimethy i-benzyl]-8-aza-bicycIo[3.2.1 ]octan-23-one oxime
- Nortropinone prepared essentially as described by Berdini et a!. (2002) Tetrahedron 58:5669; 1.50 g, 12.0 mmol
- 2,3-dimethyI-4-ethoxy(2-methoxy)benzaIdehyde (2.71 g, 13.0 mmol) are dissolved in dry CH 2 Cl? (100 mL).
- Sodium triacetoxyborohydride (3.56 g, 17,0 mmol) is added and the mixture is stirred overnight. The reaction is quenched by the addition of 1 N NaOH solution (50 mL) and the resulting layers are separated.
- the aqueous iayer is extracted with CH 2 Cl 2 (2 x 50 mL), and the combined organic layers are dried over Na 2 SC ⁇ and evaporated to yield crude intermediate.
- the crude intermediate is dissolved in pyridine (20 mL), and water (10 mL) and hydroxylamine hydrochloride (0.92 g, 13.3 mmol) are added. The solution is stirred for 6 h. The volatiles are evaporated and the product is partitioned between 1 N NaOH (50 mL) and EtOAc (50 mL), The layers are separated, and the aqueous layer is extracted with EtOAc (2 x 25 mL). The combined organic layers are washed with brine, dried over Na 2 SO 4 and evaporated.
- Step 2 8-[4-(2-Methoxy-ethoxy)-2,3-dimethy l-benzyl]-8-aza-bicycIo[3.2.1]oct-3-ylamine
- This compound is prepared a described for Compound 1, with readily apparent modification of starting materials.
- N-(8- ⁇ 4-[(R)-2-(fer/-butyl-dimethyl-silanyloxy)-propoxy]-2,3-dimethyl-benzyl ⁇ -8-aza- bicyclo[3.2.1]oct-3-yl)-3-chloro-4-fluoro-benzamide (0.06 g, 0.15 mmoi) is dissolved in dry THF (10 ml), cooled to 0 0 C, and TBAF (IM in THF, 0.15 mL, 0.15 mmoi) is added. The solution is allowed to warm to rt, stirred for a further 3 h, and the volatiles are evaporated.
- Step 1 l -(4-methoxy-2,3-dimethyiphenyl)ethanone
- the light pink solid is collected via filtration and washed with water.
- the solid is redissolved in EtOAc (500 mL), washed with water and brine, and dried over Na 2 SO 4 . Removal of the solvent under reduced pressure affords the title compound as a light pink solid.
- the organic layer from the filtration of the solid is separated, and the aqueous phase is extracted with CH 2 Cl 2 (2 x 100 mL). Organic layers are combined, washed with water (2 x 250 mL), brine (250 mL), dried over Na 2 SO 4 , and concentrated under reduced pressure. The residue is triturated with CH 2 CWEt 2 O (1 : 1, 50 mL) to afford additional title compound as a light pink solid.
- the reaction is quenched by addition of aq. NaOH (IN, 75 mL), and stirred at rt for 0.5 h.
- Insoluble materials are removed by filtration through CeI ite ® , and the filter cake is washed with EtOAc.
- the filtrate and wash are combined and concentrated under reduced pressure.
- the residue is partitioned between water and EtOAc, The organic layer is washed with water and brine, dried over Na 2 SO 4 , and concentrated under reduced pressure to afford a yellow oil.
- the residue is purified by column chromatography on silica gel (4: 1 Hexane: EtOAc).
- N-tl-l l- ⁇ -fallyloxy ⁇ -dimethylphenyljethylJpiperidin ⁇ -yl ⁇ -chloronicotinamide (100 mg, 0.234 mmol) is dissolved in xylenes, treated with r ⁇ -propylamine (40 ⁇ L, 0.468 mmol, 2.0 eq.) and heated in a sealed tube in a 180 0 C oil bath for 72 h. The solution is concentrated in vacuo and the residue purified by PTLC (2 mm silica gei plate) eluting with 4% MeOH (2M NH 3 )/DCM to provide the title compound as a tan oil.
- LCMS: T R 2.10 min, m/z 451.17 (M+l).
- Step 1 1 -[ 1 -(4-methoxy-2,3-dimethylphenyI)ethyl]piperidin-4-amine
- the reaction is quenched by addition of aq. NaOH (IN, 75 mL), and stirred at rt for 0.5 h.
- Insoluble materials are removed by filtration through Celite ® , and the filter cake is washed with EtOAc.
- the filtrate and wash are combined and concentrated under reduced pressure.
- the residue is partitioned between water and EtOAc.
- the organic layer is washed with water and brine, dried over Na 2 SO 4 , and concentrated under reduced pressure to afford a yellow oil.
- the oil is dissolved in EtOAc (20 mL), and treated with 4M HCI in dioxane (15 mL) at rt overnight. The white solid is collected via filtration, and washed with EtOAc.
- 2,5-Dichloronicotinoyl chloride is prepared from 2,5-dichloronicotinoyl acid (1.77 g, 9.22 mmol) by treatment with thionyl chloride (3.35 mL, 46.1 mmol, 5 eq.) at reflux for 18 h.
- the material is concentrated in vacuo, and used as is to acylate l-[l-(4-methoxy-2,3- dimethylphenyl)ethyl]piperidin-4-amine (900 mg, 3.43 mmol) under Schotten-Baumann conditions (ziO-propyl acetate, aqueous Na 2 CO 3 ).
- 2,5-DichIoro-N- ⁇ l-[l-(4-methoxy-2,3-dimethylphenyl)ethyl]piperidin-4-yl ⁇ nicotinamide (100 mg, 0.229 mmol) is dissolved in 5 mL xylenes, treated with w ⁇ -propylamine (50 ⁇ L, 0.458 mmol, 2.0 eq.) and heated in a sealed tube in a 195 0 C oil bath for 20 h. The solution is concentrated in vacuo and the residue purified by PTLC (2 mm silica gel plate) eluting with 4% MeOH (2M ⁇ H 3 )/DCM to provide the titie compound as a tan foam.
- 3-Chloroisonicotinoyl chloride is prepared from 3-chloroisonicotinoyI acid (1.93 g, 12.25 mmol) by treatment with thionyl chloride (4.45 mL, 61.24 mmol, 5 eq.) at reflux for 18 h.
- the material is concentrated in vacuo, and used as is to acylate I-[l-(4-methoxy-2,3- dimethylpheny!)ethyl] ⁇ i ⁇ eridin-4-amine (280 mg, 1.07 mmol) under Schotten-Baumarm conditions (/s ⁇ -propyl acetate, aqueous Na 2 CO 3 ).
- Step 1 ] -[4-(2-methoxy-ethoxy)-2,3-dimethyI-phenyl]-ethanone
- reaction mixture After stirring for 60 min at 0 0 C, the reaction mixture is poured onto 300g of ice cubes, and vigorously stirred as cone. HCI (5 mL) is added slowly. After 1 h stirring, the organic layer is isolated, washed with brine, and dried over NaiSCV Removal of the solvent under reduced pressure affords the title compound as an off white oil which becomes white crystalline after storage in a refrigerator overnight.
- Step 2 ((R)-I - ⁇ l-[4-(2-Methoxy-ethoxy)-2,3-dimethyl-phenyl]-ethyI ⁇ -pyrrolidin-2- ylmethyl)carbamicacid tert-butyl ester
- aqueous phase is made basic (pH>10) with 10 N NaOH and extracted with CH 2 CI 2 (6 x 25 ml). The combined organic layers are washed with brine, dried and concentrated to give crude product, which is used in the next step without further purification.
- LCMS: T R 1.98 min, m/z 307.09 (M+l).
- Step 4 N-((R)-l- ⁇ l-[4-(2-Methoxy-ethoxy)-2,3-dimethyl-phenyl]-ethyI ⁇ - ⁇ yrroIidin-2-ylmethyl)-6- trifluoromelhyl-nicotinamide
- the mixture is cooled to 0 0 C and 10 ml 6% NaOH is added to quench the reaction. After stirring at 0 0 C for another 30 min, the solid is removed by filtering through Celite ® . The solvent is removed under vacuum, and the residue is diluted with 50 ml H 2 O and made acidic with 6N HCI. The mixture is then extracted with ether (3 x 40 ml) to remove impurities. The aqueous phase is made basic with I O N NaOH and extracted with CH 2 CI 2 (4 x 50 ml). The combined organic layers are washed with water (30 m!), dried and concentrated to give the title compound.
- Step 7 N-((S)-l- ⁇ l-[4 ⁇ 2-Metlioxy-ethoxy)-2.3-dimethyl- ⁇ henyl]-ethyl ⁇ -pyrrolidin-2-yimethyl)-6- trifluoromethyl-nicotinamide
- Step 1 ⁇ (R)-l-[4-(2-Methoxy-ethoxy)-2,3-dimethyl-benzyl]-pyrrolidin-2-ylmethyl ⁇ -carbamic acid fert-buty ⁇ ester
- N-[l-(4-Methoxy-2,3-dimethyl-benzyl)-piperidin-2-ylmethyI]-3,4-dimethyI-benzamide (570 mg, 1.45 mrnol) in CH 2 Cl 2 (20 ml) is cooled to -78 0 C and BBr 3 (I M in CH 2 Cl 2 ; 7.9 m ⁇ j is added dropwise. The resulting mixture is stirred under a nitrogen atmosphere at -78 0 C to rt overnight.
- Step 5 rac-N- ⁇ l-[4-(4-Chloro-butoxy)-2,3-dimethyi-benzyl]- ⁇ i ⁇ eridin-2-ylmethyl ⁇ -3,4-dimethyl- benzamide and N- ⁇ 1 -[4-(4-Iodo-butoxy)-2,3-dimethyI-benzyl]-piperidin-2-ylmethyl ⁇ -3,4-dimethyl- benzamide
- a 25 nil sealed tube is charged with a mixture of rac-N- ⁇ l -[4-(4-chloro-butoxy)-2,3- dimetliyl-benzyi]-piperidin-2-ylmethyl ⁇ -3,4-dimethyl-benzamide and N- ⁇ l-[4-(4-iodo-butoxy)-2,3- dimethyl-benzyl]-piperidin-2-ylmethyl ⁇ -3,4-dimethyl-benzamide (0.37 mmol), CH 3 CN (6 ml), KI (10 mg), Cs 2 CO 3 (300 mg) and neat NMe 2 (2 ml). The resulting mixture is heated to 90 0 C for 18 h, and is then allowed to cool to rt.
- Step 1 -(l -(4-inethoxy-2.3-dimethy]pheny])ethyl)piperidin-4-amine
- reaction mixture is diluted with CH 2 Cl 2 , washed with IN NaOH and brine, dried over Na 2 SO 4 , and concentrated under reduced pressure, and the residue is purified by flash chromatography (EtOAc) to afford the title compound as an off white solid.
- reaction mixture is cooled to 0 0C, and quenched with MeOH.
- the solvent is removed under reduced pressure.
- the residue is partitioned between EtOAc and 14 saturated NaHC ⁇ 3, and the aqueous layer is extracted with EtOAc. Organic layers are combined, washed with brine, dried over Na 2 SO 4 and concentrated under reduced pressure to afford the title compound as a yellow solid.
- the reaction mixture is diluted with CH 2 Cl 2- washed with Vz saturated NaHCO 3 and brine, dried over Na 2 SOjJ, and concentrated under reduced pressure.
- the residue is purified by PTLC (hexane/EtOAc/TEA: 50/50/1) to afford a yellow oil.
- PTLC hexane/EtOAc/TEA: 50/50/1
- Zn(CN) 2 29.4 mg, 0.25 mmol
- Pd 2 (dba) 3 2.8 mg, 0.005 mmol
- DPPF 4.5 mg, 0.005 mmoi
- 2,3-Dimethylisonicotinic acid ethyl ester (4.0 g, 22.3 mmol) is dissolved in anhydrous THF (100 mL) at 0 0 C and treated with 1.5 equivalents of lithium aluminum hydride (33.5 mmol, IM solution in THF) for 1 h.
- the flask is placed in an ice-water bath and the reaction is quenched by sequential addition of water (1.3 mL), 15% NaOH in water (1.3 mL), water (1.3 mL) and 13 g of MgSO 4 .
- the resulting suspension is stirred at rt for 30 min, and filtered through Celite ® , and the filter cake is washed with 5% MeOH in DCM.
- Step 1 /e/7-Butyl (l- ⁇ l-[4-(allyloxy)-2,3-dimethylphe ⁇ yi]ethyl ⁇ yrrolidin-3-yl)carbamate
- tert-Butyl (l- ⁇ l-[4-(allyloxy)-2,3-diniethyIphenyI]ethyl ⁇ pyi ⁇ olidin-3-yl)carbamate (7.46 g, 19.9 mmol) is dissolved in EtOAc (30 mL) and treated with 4 M HCi in d ⁇ oxane (30 mL) at it overnight. The reaction mixture is then triturated with hexane, and resulting yellow solid is collected via filtration and washed with Et 2 O to afford the title compound.
- N- ⁇ l-[l-(4-hydiOxy-2,3-dimethyl ⁇ henyI)ethyl]pynOlidi ⁇ -3-yl ⁇ -3,5-dimethoxybenzamide is alkylated with any variety of agents to generate compounds of the formula:
- This compound is prepared from l-(4-melhoxy-2,3-dimethylphenyi)ethanone (5.02 g, 28.2 mmol) and (3R)-(+)-3-(/er/-butoxycarbonylamino)pyrrolidine (5 g, 26.85 mmol) via the synthetic sequence described above for l- ⁇ l-[4-(al!yloxy)-2,3-dimethylphenyl]ethyl ⁇ pyrrolidin-3-amine.
- the resulting HCl salt is partitioned between EtOAc and IN NaOH. The organic phase is washed with brine, dried over Na 2 SO 4 , and concentrated under reduced pressure to afford the title compound as a brown oil. !
- This compound is prepared from 3-hydroxy-5-methoxy-N- ⁇ l-[l-(4-methoxy-2,3- dimethylphenyl)ethyl]pyrrolidin-3-yl ⁇ benzamide as described above for Compound 31.
- This compound is prepared from 3-hydroxy-5 ⁇ methoxy-N- ⁇ l-[l-(4-rnethoxy-2,3- dimethylphenyl)ethyl]pyrrolidin-3-yI ⁇ benzamide as described above for Compound 31.
- Step 2A Preparation of(R)-l-(2,3-Dimethyl-phenoxy)-propan-2-ol
- Step 2B Preparation of4-((R)-2-Hydroxy-propoxy)-2,3-dimethyl-benzaldehyde
- Step 2C Preparation of 2-methy ⁇ -propane-2-s ⁇ dfmic acid l-[4-((R)-2-hydroxy-propoxy)-2,3- dimethyl-phenyl]-meth-(E)-ylideneamide To a mixture of 4-((R)-2-hydroxy-propoxy)-2,3-dimethyl-benzaldehyde (7.9 g. 38 mmol) in
- Step 2D Preparation of 2-methy ⁇ -propane-2-sulfinic acid ⁇ (S)-l-[4-((R)-2-hydroxy-propoxy)-2,3- dimethyl-phenyl]-ethyl ⁇ -amide
- Step 2E Preparation of (R)-I -[4- ((S)-I -amino-ethyl)-2, 3-dimethyI-phenoxy]-propan-2-ol
- Crude 2-methyl-propane-2-sulf ⁇ nic acid ⁇ (S)-l-[4-((R)-2-hydroxy-p]Opoxy)-2,3-dimethyl- phenyl]-ethyl ⁇ -amide (35g) is dissolved in 400 ml MeOH, and 100 ml 4N HCl/dioxane is added and the mixture is stirred at rt for 2 h.
- Step 4 fe?-/-butyl ⁇ (3R)-l-[(l S)-l-(4- ⁇ [(2R)-2-hydroxy ⁇ ropyI]oxy ⁇ -2,3-dimethylpheny])ethyl]-2- oxo ⁇ yrrolidin-3-yl ⁇ carbamate
- 2,3-dimethyl ⁇ henyl)ethyl]amino ⁇ butanoate (0.72 g, 1.64 mmol) is treated with aq. IN NaOH (8.2 mL) and EtOH (8.2 mL) at it overnight EtOH is removed under reduced pressure. The residue is diluted with water (5 mL), and pH is adjusted to 5-6. The white precipitates are collected, washed with water, and dried by evaporation with toluene under reduced pressure. The white solid is dissolved in DMA (20 mL), CH 2 Cl, (10 mL) and TEA (2 mL). BOP (0.82 g, 1.85 mmol) is added.
- This intermediate (21.2 g, 0.1 17 mol, 1 eq.) is dissolved in CH 2 Cl 2 (150 mL) and is slowiy added to a stirring solution of 1.0M TiCl 4 (234 mL, 0.234 mol, 2 eq.) and Cl 2 CHOCH 3 (1 1.5 mL, 0.128 mol, 1.1 eq.) cooled to - 78°C.
- the reaction is allowed to slowly warm to rt and stirred overnight.
- the reaction is poured into ice (75 g) and cone. HCl (30 mL).
- the organic layer is separated, washed with water (100 mL) and brine (100 mL), dried over Na 2 SO.;, filtered and concentrated under reduced pressure.
- This compound is prepared using the method described for Compound 31 , by reaction of I - (4-(2-methoxyelhoxy)-2,3-dimethylbenzyl)piperidin-4-amine with 2-chloroisonicotinic acid, m/z 446.1 1 (M+l).
- the reaction is cooled to rt, quenched with NaOH (IN, 10 mL) and extracted with 10 mL EtOAc. The organic layer is separated, washed with brine, dried over Na 2 SO 4 , and concentrated under reduced pressure. The residue is purified by PTLC to afford the desired intermediate as an oil, which is dissolved in anhydrous CH 3 CN (1 mL) with K 2 CO 3 (0.027 g, 0.199 mmol, 2.5 eq.). Morpholine (0.008 g, 0.096 mmol, 1.2 eq.) is added and the reaction is heated at 80 0 C overnight.
- the reaction is allowed to cool to rt and is diluted with EtOAc (5 mL) and washed with NaOH (IN, 2 x 5 mL). The organic layer is separated, washed with brine, dried over Na 2 SO 4 , and concentrated under reduced pressure. The residue is purified by PTLC to afford the title compound.
- Step 1 l-(l-(4-methoxy-2,3-dimethylphenyl)ethyl)azetidin-3-arnine
- the resulting slurry is filtered through Celite ® , and the filter cake is washed with EtOAc. The filtrate and wash are combined and concentrated under reduced pressure.
- the residue is partitioned between water and EtOAc, and the organic layer is washed with water and brine, dried over Na 2 SO.*, and concentrated under reduced pressure.
- the residue is purified by PTLC (hexane/EtOAc: 3/2) to afford a yellow oil.
- the oil is dissolved in CH 2 CI 2 (3 mL), and treated with TFA (0.5 mL) at rt overnight. Solvent is removed under reduced pressure.
- the residue is partitioned between EtOAc and IN NaOH.
- the title compound is prepared from l-(l-(4-methoxy-2,3-dimethy]phenyI)ethyI)azetidin-3- ami ⁇ e and 3-chloro-4-fluoro-benzoic acid essentially as described for Compound 51.
- the title compound is prepared from l-(l-(4-methoxy-2,3-dimethylphenyl)ethyl)azetidin-3- amine and 3-(2-fIuorophenyl)-arcylic acid essentially as described for Compound 51.
- the mixture is stirred at 0 0 C for 1 h and then at rt overnight.
- the reaction is poured into a flask containing 70 mL Na 2 CO 3 (minimal THF to transfer).
- the reaction is stirred at rt for 1 h, after which time 80 mL of diethyl ether is added.
- the biphasic solution is allowed to stir vigorously at it for 30 min.
- the organic layer is decanted off the white precipitate.
- the white solid is washed an additional two times with 80 mL diethyl ether in the same manner.
- Step 3 4- ⁇ l-[4-(3-Chloro-4-fIuoro-benzoylamino)-piperidin-l-yl]-ethyl ⁇ -3-methyl-benzoic acid isopropyl ester
- Step 5 4-(l - ⁇ 4-[(3-chloro-4-fluorobenzoyl)ammo]piperidin-] -yl ⁇ ethyl)-N-(2-methoxyethyl)-N,3- dimethylbenzamide
- n 0 or L
- n 1 and R 20 is piperidine
- This intermediate is prepared using N-(2-aminoethyl)piperidinc via the procedure described above for intermediate I ⁇ l .
- ⁇ is 1 and R 2 o is pyrrolidine
- This intermediate is prepared using 4-(2-aminoethyI)morpholine via the procedure described above for intermediate 1-1.
- n 1-5.
- R 20 is N(CH 3 ),
- This intermediate is prepared using N,N-dimelhy (ethylene diamine via the procedure described above for intermediate I- 1. 1-6. n is 1 and R 20 is CH 2 N(CHa) 2
- This intermediate is prepared using N,N-dimethyl-l,3-propane diamine via the procedure described above for intermediate 1-1.
- n 1 and R 20 is CH 2 OCH 3
- This intermediate is prepared using 3 -methoxypropy famine via the procedure described above for intermediate 1-1.
- N-alkylated intermediates 1-1 to 1-7 are acylated with any variety of agents to generate acylated intermediates of the Formula:
- n 0 or 1.
- n 1 and R 2 O is piperidine
- ⁇ is 1 and R 20 is CH 2 N(CH 3 ) 2
- n 0 or 1.
- n 1-18.
- R 2 o is morpholine
- n 1-20.
- R 20 is CH 2 N(CH 3 ),
- This intermediate is prepared from 1-6, via the procedure described above for 1-15, LCMS: m/z 350.10 (M+l).
- ⁇ is 1 and R 20 is CH 2 OCH 3
Abstract
Aminopiperidines and related compounds of the formulas are provided: in which variables are described herein. Such compounds may be used to modulate MCH receptor activity in vivo or in vitro, and are particularly useful in the treatment of a variety of metabolic, feeding and sexual disorders in humans, domesticated companion animals and livestock animals. Pharmaceutical compositions and methods for treating such disorders are provided, as are methods for using such Iigands for detecting MCH receptors (e.g., receptor localization studies).
Description
AMINOPIPERIDINES AND RELATED COMPOUNDS
FIELD OF THE INVENTION
This invention relates generally to aminopiperidines and related compounds. The invention further relates to the use of such compounds for treating a variety of metabolic, eating and sexual disorders, and as probes for the detection and localization of melanin concentrating hormone receptors.
CROSS-REFERENCE TO RELATED APPLICATIONS This application claims priority to U.S. Provisional Application 60/820,834, filed July 31,
2006, which is hereby incorporated by reference in its entirety.
BACKGROUND OF THE INVENTION
Melanin concentrating hormone, or MCH, is a cyclic 19 amino acid neuropeptide first identified as a regulator of skin coloration in fish and other vertebrates, and subsequently as a regulator of food intake and energy balance in higher vertebrates. The activity of this hormone in the body is mediated via binding to specific G protein-coupled receptors, including Melanin Concentrating Hormone Receptor- 1 (MCHlR) and Melanin Concentrating Hormone Receptor-2 (MCH2R). Certain monkey and human MCHlR and MCH2R sequences, as well as various chimeric MCH receptor proteins, have been disclosed in U.S. Patent Application Publication Number 2003/0148457, 2003/0166834 and U.S. Patent Number 7,078,484.
Agents capable of modulating MCH receptor activity are highly desirable for the treatment of a variety of diseases and disorders, including obesity, metabolic syndrome, eating disorders (e.g., bulimia and anorexia), sexual disorders (e.g., anorgasmic or psychogenic impotence) and metabolic disorders, such as diabetes. Small molecule, non-peptide antagonists of MCH receptors would be of particular value for such therapies. The present invention fulfills this need, and provides further related advantages.
SUMMARY OF THE INVENTION The present invention provides aminopiperidines and related compounds of Formula 1:
Formula I

RA is 6- to IO-membered aryi, 5- to I O-membered heteroaryl, Q-Csalkyl, Cj-Cehaloaikyl, (C3- Cgcycloalkyl)Co-Gia]kyl or (5- to 7-membered heterocycloalkyl)Co-QalkyI, each of which is optionally substituted and each of which is preferably substituted with from 0 to 4 substituents independently chosen from RB; Each RB is independently:
(i) halogen, hydroxy, nitro, cyano, amino, -COOH, oxo, amiπocarboπyl or aminosulfonyi; (ii) CrC6alkyl, C2-C6alkenyl, C2-C6alkynyL C,-C6a]koxy, CrC6alkanoyl, CrC6alkylthio, C,-
Qalkanoyloxy, CpQalkoxycarbonyl, Ci-Csalkylsulfonyl, Cj-Cήhaloalkyl, mono- or di-(C|-
C(,alkyl)aminoCo-C4alkyl, mono- or di-(Ci-C6alkyl)aminocarbonylCo-C4a!kyl, mono- or di-
(C1-C6alkyl)aminosuIfonylC0-Cialkyi, (C3-C7cycIoaIkyl)C0-C2alkyl, (4- to 10-membered heteiOcyc]e)Co-C2alkyl, phenylCo-C2alkyl or (3- to 10-membered cycIe)Co-C2alkoxy; each of which is optionally substituted and each of which is preferably substituted with from O to
4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo,
 d-Cjalkoxy, C]-C4haloalkyl, C!-C4haloalkoxy, mono- or di-(C]-C6alkyl)amino and 5- or 6- membered heterocycloalkyl; or
d-Cjalkoxy, C]-C4haloalkyl, C!-C4haloalkoxy, mono- or di-(C]-C6alkyl)amino and 5- or 6- membered heterocycloalkyl; or
(iii) taken together with a R1 1 or R!2 moiety to form a C5-C7cycloaikyl or a 5- to 7-membered heterocycloalkyl; or two adjacent RB groups are taken together to form a 5- or 6-membered carbocycle or a 5- or 6-membered heterocycle, each of which is optionally substituted and each of which is preferably substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Q^alkyi, Cj-C4alkoxy, Ci-C4haloalkyl, and mono- or di-(Cp C6alkyl)amino;

U and T are independently N or CRy; P is N or CRi5; Q is N or CR9; Ri is cyano, nitro, halogen or a group of the formula -L-M, wherein:
L is a single covalent bond, O, C(=0), OC(O), C(O)O, OC(O)O, S(0)w, N(Rx), C(O)N(Rx), NCRJC(O), N(Rx)S(O)1,, or S(O)wN(Rλ), wherein each Rx is independently
hydrogen, Cj-Csalkyl, C^-Cealkenyl, C2-Cealkynyl or Ci-Cehaloalkyl and w is independently selected at each occurrence from 0, I and 2; and
M is hydrogen, Q-CgalkyL C2-C8alkenyl, C2-C8aikynyl, C2-C8alkyl ether, mono- or di-(Q-
CsaIkyl)aminoC0-C6aikyI, (C3-C7cycloalkyi)C0-C6aikyl or (5- to 10-membered heterocycle)CcrC6alkyi, each of which is optionally substituted and each of which is preferably substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imiπo, aminosulfonyl, -COOH, cyanoimido,
C,-C5alkyl, Ci-C6haloalky1, C,-C6alkoxy, CrC6haϊoalkoxy, CrC6aIkylthio, C1-
C6aikylsuifonyl, CrQalkanoyl, CrC6alkanoyloxy, CrC6alkanoyiamino, C1- Cgalkylaminosulfonyl, Ci-Cealkoxycarbonyl, and mono- and di-(C|-C6alkyl)aminoCo-
Qalkyl; or R] is taken together with R9 to form a fused C5-C3cycloalkyi or 5- to 8-membered heterocycloalkyl; in certain embodiments, R1 is not H;
R2 represents from 0 to 4 substituents independently chosen from Q -Chalky], CrC4haloalkyl and oxo; or two R2 groups are taken together to form a bridge (e.g., methylene, ethylene, propylene or -CH2-O-CH2-);
R3 is hydrogen, C]-C4aikyi or Ci^halσalkyl; R4 is:
(i) hydrogen; or (ii) CpCgalky! or C2-C8alkenyl, each of which is substituted with from 0 to 2 substituents independently chosen from hydroxy, amino, oxo, Ci-Cgalkoxy, CrC6alkanoyloxy, mono- or di-(CrC6alky])amino, mono- or di-(CrC6alkyl)aminocarbonyl and (4- to 7-membered heteiOcycloaIkyI)Co-C2alkyl;
Each R7, R9 and R15 is independently hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH. aminocarbonyl. aminosulfonyi, CrQalkyl. C2-Qalkenyl, C2-C6alkynyl, C]-C6alkoxy, Cr
Cfialkanoyl, Ci-Cήalkanone, Ci-Cβalkanoyloxy, C2-C6alkyl ether, CrCsalkoxycarbonyi, Cr
Cβaikylthio, Q-Cealkylsulfonyl, mono- or di-(Ci-C6aIkyl)aminoC0-Cjalkyl, mono- or di-(Cj~
CealkyOaminocarbonylCo-C^aikyl, mono- or dHC]-Qa!kyl)aminosu!fonylCo-QaikyL C5-
C6haloaikyl, Cj-C6haloalkoxy, Ci-Cehydroxyalkyi, CpCsaminoaikyl, CrCgcyanoalkyl, or mono- or di-(CrC6alkyl)aminoC0-C4alkyl;
Or R9 is taken together with R; to form a fused cycloalkyl or heterocycloalkyl; and RI G is hydrogen, Q-Cgalkyl, C^-Cealkenyl or C2-C6alkynyL
Certain aminopiperidines and related compounds of Formula I further satisfy Formula II:
Formula II

Z is O, NR14, C(=O)N(Ru) or N(R14)C(O), wherein RM is H or d-Qalkyl;
Rs is hydrogen, Q-Cgalkyl, C2-Cgaikenyl, Cj-CgaJkynyl. C2-C3alkyl ether, mono- or di-(Cr CgalkyOaminoCo-Cήalkyl, (C3-C7cycloalky])Co-C6alkyl or (5- to 10-membered heteiOcycle)Co-Csalkyl, each of which is optionally substituted and each of which is preferably substituted with from O to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosulfonyl, -COOH, C,-C6alkyl,
C]-C6alkoxy, Q-Cβhaloalkyl, C]-C6haloalkoxy, mono- or di-(Ci-C6alkyl)aminoCø-Qalkyl, CrC6aϊkylsulfonyI, CrQalkylthio. Cj-Qsalkylaminosulfonyl, Cj-C6a!koxycarbonyl or Cr
Cgalkanoylamino;
R9 and R!5 are independently halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl, aminosulfonyl, CrQalkyl, C2-Qalkenyl, C2-Qalkynyl. Cj-Qalkoxy, CpCealkanoyl, C^- C6alkanone, Ci-C6alkanoyloxy, C2-C6alky} ether, C]-C6alkoxycarbonyl, Ci-Csalkylthio. Q- Csalkylsulfonyl. mono- or di-(C}-C6alkyl)aminoC0-C4alkyl, mono- or di-(Ci-
C6alkyl)aminocarbonylC0-C4alkyl, mono- or di-(C]-C6alkyl)aminosulfonylCo-C4alkyl, C1- C6haloalkyl, Ci-Cghaloalkoxy, Cj-Cghydroxyalkyl, Ci-Cβaminoalkyl, CrC6cyanoalkyl, or mono- or di-(CrCfialkyl)aminoC0-C4alkyl; Or R8 is taken together with R9 to form a fused heterocycloalkyl; and the remaining variables are as described for Formula I.
Stilt further aminopiperidines and related compounds of Formula 1 satisfy Formula IJI:
Formula III
 wherein R2 represents from 1 to 4 substituents independently chosen from Q^alkyl, Cr
wherein R2 represents from 1 to 4 substituents independently chosen from Q^alkyl, Cr
C4haloalkyi and oxo; and the remaining variables are as described for Formula I.
Further aminopiperidin.es and related compounds of Formula I also satisfy Formula IV:
V
wherein:

P is N, CH or CR15;
Ri is a group of the formula -L-M, wherein: L is a single covalent bond, O, C(O)8 OC(O), C(O)O, OC(O)O, S(0)w, N(Rx),
C(O)N(Rx), N(Rx)C(O), N(Rx)S(O)W, or S(O)WN(RX), wherein each Rx is independently hydrogen, C,-C6alkyl, C2-C6aikenyl. C2-C6alkynyl or CrC6haloalkyl and w is independently selected at each occurrence from O, 1 and 2;
M is hydrogen, Ci-C3alkyi, Ci-Cgalkenyl, Cϊ-Cgalkynyl, Ca-Cgalkyl ether, mono- or di-(Cr C8aiky])aminoC0-C6alkyl, (C3-C7cycloalkyi)Co-C6aikyI or (5- to 10-membered heterocycle)Co-C6aikyl, each of which is substituted with from O to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarboπyl, imino, aminosulfonyl, -COOH, cyanoimido, CrQalkyl, Ci-Cghaloalkyl, Ci-Cfialkoxy, Cr
C6haloalkoxy, CrC6alkylthio, Ci-C6alkyIsulfonyL Ci-C6alkanoyi, C]-C6alkanoyloxy, Cr Qalkanoylamino, Ci-Cήalkylaminosulfonyl, C[-C6alkoxycarbonyl, and mono- and di-(Cr
C(-,a!ky I)am i noCo-C6alky 1 ; or R] is taken together with R9 to form a fused Cs-Cgcycloalkyl or 5- to 8-membered heterocyc loal ky i ; such that R] is not hydrogen; R9 and R|5 are independently halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl, aminosulfonyl. Cj-Cealkyi, C2-C6alkenyl, C^-Cήalkynyl, C]-C6alkanoyl, Cs-Qalkanone, C;- C6aϊkanoy!oxy, C2-C5alkyi ether, CrC6alkoxycarbonyl, C]-Cealkylthio; Cs-C^alkylsulfonyl, mono- or di-(Ci-Cόalkyl)ammoCo-C4alkyl, mono- or di»(CrCήa!kyl)aminocarbonylCo-C4afkyl, mono- or di-fCrQalkylJaminosuifoπyiCo-Cjafkyl, CpCehaloalkyl, Ci-Cehaloalkoxy, Cp Cβhydroxyalkyl, Ci-C6aminoalkyl, Ci-C6cyanoalkyl, or mono- or di-(CrC6a]kyi)ammoC0-
Cj alky 1; or R9 is taken together with R: to form a fused cycloalkyl or heterocycloalkyl; and the remaining variables are as described for Formula I.
Other aminopiperidines and related compounds of Formula I further satisfy Formula V, VI, or VII or VIIl:

Formula V Formula VI

Formula VII Formula VIII or are a pharmaceutically acceptable salt, solvate or esters of such a compound. Variables within Formulas V, VI3 VIl and VIII are generally as described for Formula I.
Further aminopiperidines and related compounds provided herein satisfy Formula IX.
Formula IX
 or are a pharmaceutically acceptable salt, solvate or esters of such a compound. Variables within
or are a pharmaceutically acceptable salt, solvate or esters of such a compound. Variables within
Formula TX are generally as described for Formula I.
Within certain aspects, aminopiperidines and related compounds provided herein are MCH receptor modulators and exhibit a K1 of no greater than 1 micromolar, 500 nanomolar, 100 nanomolar, or 10 nanomolar in a MCH receptor binding assay and/or have an ECj0 or IC5O value of no greater than 1 micromolar, 500 nanomolar, 100 nanomolar, or 10 nanomolar in an assay for determining MCH receptor agonist or antagonist activity.
Within certain aspects, aminopiperidines and related compounds provided herein are labeled with a detectable marker (e.g., radiolabeled or fluorescein conjugated). The present invention further provides, within other aspects, pharmaceutical compositions comprising at least one aminopiperidine or related compound provided herein in combination with a physiologically acceptable carrier or excipient. Within certain embodiments, a pharmaceutical composition provided herein may further comprise one or more additional active agents (i.e., drugs).
Pharmaceutical compositions provided herein may be formulated, for example, as an injectable fluid, an aerosol, a cream, an oral liquid, a tablet, a gel, a pill, a capsule, a syrup or a transdermal patch.
Methods are further provided for modulating binding of ligand (e.g., MCH) to cellular MCH receptor, comprising contacting cells expressing MCH receptor with a MCH receptor modulator as described above, in an amount that would be sufficient to detectabϊy modulate MCH binding to MCH receptor in vitro. The cells may, but need not, be present in a human nor non-human animal. 5 Tn other aspects, methods are provided for modulating binding of ligand (e.g., MCH) to
MCH receptor in vitro, comprising contacting MCH receptor with a MCH receptor modulator as described above, in an amount sufficient to detectably modulate MCH binding to MCH receptor.
Within further aspects, the present invention provides methods for modulating the signal- transducing activity of MCH receptor in a cell, comprising contacting a cell expressing MCH I O receptor, either in vivo or in vitro, with a MCH receptor modulator as described above, under conditions and in an amount that is sufficient to detectably alter the electrophysiology of the cell. Within certain embodiments of the above methods, the MCH receptor is a MCHlR. The present invention further provides, within other aspects, methods for treating a disease or disorder associated with MCH receptor activation, comprising administering to a patient in need 5 of such treatment a therapeutically effective amount of a MCH receptor modulator as described above. Such diseases and disorders include, for example, obesity, metabolic syndrome, eating disorders (e.g., bulimia nervosa), sexual disorders, diabetes, heart disease and stroke. The MCH receptor modulator may be administered oralfy, or via another means such as intranasal Iy, intravenously or topically. Within certain embodiments, the patient is a human, companion animal0 (e.g., dog or cat) or livestock.
Also provided herein are methods for treating a patient, comprising diagnosing the patient as having a disease or disorder associated with MCH receptor activation, correlating the diagnosis of a disease or disorder associated with MCH receptor activation with the need for administration of a
MCH receptor modulator, and administering to the patient an effective amount of a MCH receptor5 modulator as described above.
Methods are provided, within other aspects, for determining the presence or absence of MCH receptor in a sample, comprising: (i) contacting a sample with a compound as described above under conditions that permit binding of the compound to MCH receptor; and (ii) detecting a level of the compound bound to MCH receptor. Within certain embodiments, the compound is radiolabeled,0 and the step of detection comprises: (i) separating unbound compound from bound compound; and (ii) determining an amount of bound compound in the sample. Detection may be achieved, for example, using autoradiography. Representative samples include, for example, tissue sections.
Packaged pharmaceutical preparations are also provided, comprising: (a) a pharmaceutical composition as described above in a container; and (b) instructions for using the composition to treat5 a patient suffering from or at risk for developing a disease or disorder associated with MCH receptor activation.
In yet another aspect, methods for preparing the compounds disclosed herein, including the intermediates, are also provided herein.
These and other aspects of the present invention will become apparent upon reference to the following detailed description.
DETAILED DESCRIPTION OF THE INVENTION
As noted above, the present invention provides aminopiperidines and related compounds of
Formula I. Within certain aspects, such compounds are MCH receptor modulators that may be used in vitro or in vivo, to inhibit MCH binding to MCH receptors, activate MCH receptors, or to otherwise modulate MCH receptor activity in a variety of contexts, as discussed in further detail below.
TERMINOLOGY
Compounds are generally described herein using standard nomenclature. For compounds having asymmetric centers, it should be understood that (unless otherwise specified) all of the optica] isomers and mixtures thereof are encompassed. In addition, compounds with carbon-carbon double bonds may occur in Z- and E- forms, with all isomeric forms of the compounds being included in the present invention unless otherwise specified. Where a compound exists in various tautomeric forms, a recited compound is not limited to any one specific tautomer, but rather is intended to encompass all tautomeric forms. Compound descriptions are intended to encompass compounds with all possible isotopes of atoms occurring in the compounds. By way of general example, and without limitation, isotopes of hydrogen include tritium and deuterium and isotopes of carbon include 11C, 13C and ]AC. Certain compounds are described herein using a genera! formula that includes variables (e.g., X, V, R3). Unless otherwise specified, each variable within such a formula is defined independently of any other variable, and any variable that occurs more than one time in a formula is defined independently at each occurrence. In general, the variables may have any definition described herein that results in a stable compound.
The term "aminopiperidines and related compounds" refers to any compound that satisfies Formula I, or other Formula provided herein, or is a pharmaceutically acceptable salt, solvate (e.g., hydrate) or ester of such a compound. Certain aminopiperidines and related compounds further satisfy one or more additional formulas provided herein. A "pharmaceutically acceptable salt'1 of a compound recited herein is an acid or base salt that is suitable for use in contact with the tissues of human beings or animals without excessive toxicity or carcinogenicity, and preferably without irritation, allergic response, or other problem or complication. Such salts include mineral and organic acid salts of basic residues such as amines, as well as alkali or organic salts of acidic residues such as carboxylic acids. Specific pharmaceutically acceptable anions for use in salt formation include, but are not limited to, acetate, 2- acetoxybenzoate, ascorbate, benzoate, bicarbonate, bromide, calcium edetate, carbonate, chloride,
citrate, dihydrochloride, diphosphate, ditartrate, edetate, estolate (ethylsuccinate). formate, fumarate, gluceptate. gluconate, glutamate, glycolate, glycollylarsanilate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroiodide, hydroxymaieate, hydroxynaphthoate, iodide, isethionate, lactate, lactobionate, malate, maleate, mandelate, methylbroraide, methylnitrate, methylsulfate, mucate, napsylate, nitrate, pamoate, pantothenate, phenylacetate, phosphate, polygalacturonate, propionate, salicylate, stearate, subacetate, succinate, sulfamate, sulfanilate, sulfate, sulfonates including besylate (benzenesuifonate). carnsylate (camphorsulfonate), edisylate (ethane- 1 ,2- disulfonate), esylate (ethanesulfonate) 2-hydroxy ethyl sulfonate, mesylate (methanesulfonate), Inflate (trifluoromethanesulfonate) and tosylale (p-toluenesulfonate), tannate, tartrate, teoclate and trieth iodide. Similarly, pharmaceutically acceptable cations for use in salt formation include, but are not limited to ammonium, benzathine, chloroprocaine, choline, diethanolamine, ethylenediamiπe, meglumine, procaine, and metals such as aluminum, calcium, lithium, magnesium, potassium, sodium and zinc. Those of ordinary skill in the art will recognize further pharmaceutically acceptable salts for the compounds provided herein. In general, a pharmaceutically acceptable acid or base salt can be synthesized from a parent compound that contains a basic or acidic moiety by any conventional chemical method. Briefly, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, the use of nonaqueous media, such as ether, ethyl acetate, ethanol, methanol, isopropanol or acetonitrile, is preferred. It will be apparent that each compound of Formula I may, but need not, be formulated as a solvate (e.g., a hydrate) or a non-covalent complex. In addition, the various crystal foπns and polymorphs are within the scope of the present invention. Also provided herein are prodrugs of the aminopiperidines and related compounds provided herein. A "prodrug" is a compound that may not fully satisfy the structural requirements of the compounds provided herein, but is modified in vivo, following administration to a patient, to produce an aminopiperidine or related compound. For example, a prodrug may be an acylated derivative of a compound as provided herein. Prodrugs include compounds wherein hydroxy, amine or sulfhydryi groups are bonded to any group that, when administered to a mammalian subject, cleaves to form a free hydroxy, amino or sulfhydryl group, respectively. Examples of prodrugs include, but are not limited to, acetate, formate, phosphate and benzoate derivatives of alcohol and amine functional groups within the compounds provided herein. Prodrugs of the compounds provided herein may be prepared by modifying functional groups present in the compounds in such a way that the modifications are cleaved in vivo to yield the parent compounds.
As used herein, the term "alkyl" refers to a straight or branched chain saturated aliphatic hydrocarbon. Alkyl groups include groups having from 1 to 8 carbon atoms (Ci-Qalkyl), from 1 to
6 carbon atoms (Ci-C6alkyl) and from 1 to 4 carbon atoms (Cj-C4alkyl), such as methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl, tert-butyi, pentyl, 2-pentyl, isopentyl, neopentyl, hexyl, 2-
hexyl, 3-hexyl and 3-methylpentyl. "C0-Cna3kyl" refers to a single covalent bond (C0) or an alky! group having from 1 Io n carbon atoms; for example, "CVCealkyi" refers to a single covalent bond or a Cj-Qalkyl group. In some instances, a substituenl of an alky! group is specifically indicated. For example, "Ci-Cehydroxyalkyl" refers to a CrCgalkyl group that has at least one hydroxy substituent; Ci-C6aminoalkyl refers to a CrC6alkyI group that has at least one amino substituent; and Ci-CέCyanoalkyi refers to a CpCsalkyl group that has at least one cyano (C≡N) substituent.
"Alkylene" refers to a divalent alkyi group, as defined above. Co-C4alkyleπe is a single covalent bond or an alkylene group having from 1 to 4 carbon atoms.
"Alkenyl" refers to straight or branched chain alkene groups, which comprise at least one unsaturated carbon-carbon double bond. Alkenyl groups include, for example, C2-C8alkenyl and
C2-Cήalkenyi groups, which have from 2 to 8 or from 2 to 6 carbon atoms, respectively {e.g., ethenyl, allyl or isopropenyl). "Aikynyl" refers to straight or branched chain alkyne groups, which have one or more unsaturated carbon-carbon bonds, at least one of which is a triple bond. Aikynyl groups include, for example, Cj-Cgalkynyl and Ci-Cgalkynyl groups, which have from 2 to 8, or from 2 to 6 carbon atoms, respectively.
A "cycloalkyl" is a group that comprises one or more saturated and/or partially saturated rings in which ail ring members are carbon, such as cyclopropyl, cyciobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, adamantyl. decahydrσ-naphthalenyl, octahydro-indenyl, and partially saturated variants of the foregoing, such as cyclohexenyl. Certain cycloalkyl groups are Ci -C7 cycloalkyl, in which the group contains from 3 to 7 ring members. A "cycloalkyiCo-Cnalkyi" is a cycloalkyl group linked via a single covalent bond or a CrCnalkylene group (e.g., (C3- C7cycloalkyl)C0-C,alkyl).
By "alkoxy," as used herein, is meant an alky I group as described above attached via an oxygen bridge. Alkoxy groups include Cj-Cβalkoxy and C|-C4alkoxy groups, which have from 1 to 6 or from 1 to 4 carbon atoms, respectively. Methoxy, ethoxy. propoxy, isopropoxy, n-butoxy, sec- butoxy. ter/-butoxy, n-pentoxy, 2-pentoxy, 3-pentoxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy. and 3-methyIpentoxy are representative alkoxy groups. Similarly, "alkylthio" refers to an alkyl group as described above attached via a sulfur bridge.
"Alkylsulfonyl" refers to groups of the formula -{Sθ2)-aikyL in which the sulfur atom is the point of attachment. Alkylsulfonyl groups include Ci-C6alkylsulfonyl and Ci-C4alkylsuifonyl groups, which have from 1 to 6 or from 1 to 4 carbon atoms, respectively. Methylsulfonyl is one representative alkylsulfonyl group.
The term "oxo" is used herein to refer to an oxygen substituent of a carbon atom that results in the formation of a carbonyl group (C=O). An oxo group that is a substituent of a nonaromatic carbon atom results in a conversion of -CH2- to -C(=O)-. An oxo group that is a substituent of an aromatic carbon atom results in a conversion of -CH- to -C(=O)- and may result in a loss of
aromaticify. Similarly, an "imino" substituent results in the conversion of -CH2- or -CH- to C=NH, and substitution with a "cyanoimido" group results in the conversion Of-CH2- or -CH- to C=N-C≡N. The term "aikanoyl" refers to an acyi group (e.g., -(C=O)-aikyI). Alkanoyl groups have the indicated number of carbon atoms, with the carbon of the keto group being included in the numbered carbon atoms. For example, a C2alkanoyl group is an acetyl group having the formula - (C=O)CH.!- Alkanoyl groups include, for example, C^-Cealkanoyl, which have from 2 to 6 carbon atoms. "Cialkanoyl" refers to -(C=O)H, which (along with C2-C6alkanoyl) is encompassed by the term "Cι-C6alkanoyi."
"Alkyl ether" refers to a linear or branched ether substituent (i.e., an alkyl group that is substituted with an alkoxy group). Such groups include C2-C3alkyi ether and C2-C6alkyl ether. A C2alkyl ether group has the structure -CH2-O-CH3.
The term "alkoxy car bony 1" refers to an alkoxy group attached through a keto (-(C=O)-) bridge (i.e., a group having the general structure -C(=O)-O— alkyl). Alkoxycarbonyl groups include, for example, Ci-Ce and C]-C4alkoxycarbonyl groups, which have from 1 to 6 or from 1 to 4 carbon atoms, respectively, in the alkyl portion of the group (i.e., the carbon of the keto bridge is not included in the indicated number of carbon atoms). "C] alkoxycarbonyl" refers to
 C3alkoxycarbonyl indicates -C(-OKHCH2)2CH3 or -C(=0)-0-(CH)(CH3)2.
C3alkoxycarbonyl indicates -C(-OKHCH2)2CH3 or -C(=0)-0-(CH)(CH3)2.
"Alkanoylamino," as used herein, refers to an alkanoyl group attached through an amino linker (i.e., a group having the general structure -N(R)-C(=O)-alkyi), in which R is hydrogen or Cr Qalkyi). Alkanoylamino groups include, for example, C2-C6alkanoylamino groups, which have from 2 to 6 carbon atoms.
"Alkylamino" refers to a secondary or tertiary amine having the general structure -NH- alkyl or -N{alkyl)(alkyl), wherein each "alkyl" is selected independently from alkyl, cycloalkyl and
(cycioaikyl)alkyl groups. Such groups include, for example, mono- and di-(C]-CgalkyI)amino groups, as well as mono- and di-(C|-C6alkyI)amino groups and mono- and di-(CrC^alkyl)amino groups.
"Alkylaminoalkyl" refers to an aikylamino group linked via an alkylene group (i.e., a group having the general structure -alkylene-NH-alkyl or -alkyIene-N(aIkyl)(alkyI), in which each alkyi is selected independently from alkyl, cycloalkyl and (cycloalkyl)alkyl groups). Alkylaminoalkyl groups include, for example, mono- and di-(Ci-Cgalkyl)aminoCrCga!kyl, mono- and di-(Cj- C6atkyl)ammoCj-C6alkyl and mono- and dHCj-CβalkyOaminoCi-Qalkyl. "Mono- or di-(Cr C6alkyl)aminoC0-C6alkyl" refers to a mono- or di-(CrC6alky!)amino group linked via a single covalent bond or a Ci-Cealkylene group. The following are representative alkyl am inoaikyl groups:

The term "aminocarbonyl" refers to an amide group {i.e., -(C=O)NH2). "Mono- or di-(Cr Cgalkyl)aminocarbonyi" is an aminocarbonyl group in which one or both of the hydrogen atoms is replaced with Ci-Cgalkyi. If both hydrogen atoms are so replaced, the alkyl groups may be the same or different. "Mono- or di-(CrC6a!kyl)aminocarbonylC0-C4alkyl is a mono- or di-(Cr
Cr,alkyi)aminocarbonyl group that is attached via a single covalent bond or a Cj-Qalkylene group.
"Aminosulfonyi" refers to groups of the formula "-(SOi)-NH2, in which the sulfur atom is the point of attachment. The term "mono- or di-(C]-Cπalkyl)aminosulfonyl" refers to groups that satisfy the formula -{SOi)--NR2, in which the sulfur atom is the point of attachment, and in which one R is CrCnalkyI and the other R is hydrogen or an independently chosen Cj-Cπalkyl. "Mono- or di-(Ci-C6alkyl)aminosulfonylCo-C4alkyl is a mono- or di-(C]-C6alkyl)aminosulfonyl group that is attached via a single covalent bond or a Q-C^alkyiene group. The term "halogen" refers to fluorine, chlorine, bromine or iodine.
A "haioalkyl" is an alkyl group that is substituted with 1 or more independently chosen halogens {e.g., "CrQhaloalkyl" groups have from 1 to 6 carbon atoms, each of which is optionally substituted with 1 or more halogens). Examples of haloalkyl groups include, but are not limited to. mono-, di- or tri-fiuoromethyl; mono-, di- or tri-chloromethyϊ; mono-, di-, tri-, tetra- or penta- fluoroethyl; mono-, di-, tri-, tetra- or penta-chloroethyl; and 1,2,2,2-tetrafiuoro-l-trifluoromethyi- ethyl. Typical haioalkyl groups are trifluoromethy! and difluoromethyl. The term "haloalkoxy" refers to a haloalkyi group as defined above attached via an oxygen bridge. "CrC6haIoalkoxy" groups have 1 to 6 carbon atoms.
A dash ("-") that is not between two letters or symbols is used to indicate a point of attachment for a substituent. For example, -CONH2 is attached through the carbon atom.
A "carbocycle" or "carbocyclic group" comprises at least one ring formed entirely by carbon-carbon bonds (referred to herein as a carbocyclic ring), and does not contain a heterocycle. Unless otherwise specified, each ring within a carbocycle may be independently saturated, partially saturated or aromatic, and is optionally substituted as indicated. A carbocycle generally has from 1 to 3 fused, pendant or spiro rings; carbocycies within certain embodiments have one ring or two fused rings. Typically, each ring contains from 3 to 8 ring members {i.e., C3-C5); C5-C7 rings are recited in certain embodiments. Carbocycies comprising fused, pendant or spiro rings typically contain from 9 to 14 ring members. Certain carbocycies are C4-C]0 (i.e., contain from 4 to 10 ring members and I or two rings). Certain representative carbocycies are cycloalkyl as described above. Other carbocycies are aiyl (i.e., contain at least one aromatic carbocyclic ring, with or without one or more additional aromatic and/or cycloalkyl rings). Such aryl carbocycies include, for example, phenyl, naphthyl (e.g., 1-naphthyl and 2-naphthyl), biphenyl, fluorenyl, indanyl and 1 ,2,3,4-
tetrahydro-naphthyl. In certain embodiments, preferred carbocycles are carbocycles having a single ring, such as phenyl and 3- to 7-membered cycloalkyl groups (C3-C7cycloalkyl).
As used herein, the term "aryl" indicates aromatic groups containing only carbon in the aromatic ring or rings. Such aromatic groups may be further substituted with carbon and/or non- carbon atoms or groups. Typical aryl groups contain 1 or 2 separate, fused, or pendant rings and from 6 to about 12 ring atoms, without heteroatoms as ring members. 6- to 10-membered aryl groups include phenyl, naphthyl and phenyl groups that are fused to a 5 to 7-membered saturated or partially saturated ring that optionally contains 1 or 2 heteroatoms independently chosen from N, O and S (e.g., 3,4-methyIenedioxy-phenyl). The term "arylalkyl" refers to an aryl group linked via an alkylene bridge. For example, phenylCo-C^alkyl indicates a phenyl group that is attached via a single covalent bond (ρhenylCoalkyi) or attached through an alkylene group having 1 or 2 carbon atoms. Similarly, an aryl group may be attached through other linker groups; such groups include, for example, arylCV C6alkanoylamino and arylalkoxy groups, in which the aryl is attached via the indicated linker group. A "heterocycle" or "heterocyclic group" has from 1 to 3 fused, pendant or spiro rings, at least one of which is a heterocyclic ring (i.e., one or more ring atoms is a heteroatom independently chosen from O, S and N, with the remaining ring atoms being carbon). Additional rings, if present, may be heterocyclic or carbocyclic. Typically, a heterocyclic ring comprises 1, 2, 3 or 4 heteroatoms; within certain embodiments each heterocyclic ring has 1 or 2 heteroatoms per ring. Each heterocyclic ring generally contains from 3 to 8 ring members (rings having from 4 or 5 to 7 ring members are recited in certain embodiments) and heterocycles comprising fused, pendant or spiro rings typically contain from 9 to 14 ring members. Certain heterocycles comprise a sulfur atom as a ring member; in certain embodiments, the sulfur atom is oxidized to SO or SO2. Heterocycles may be optionally substituted with a variety of substituents, as indicated. Unless otherwise specified, a heterocycle may be a heterocycloalkyl group (i.e., each ring is saturated or partially saturated) or a heteroaryl group (i.e., at least one heterocyclic ring within the group is aromatic), such as a 5- to 10-membered heteroaryl (which may be monocyclic or bicyclic) or a 6- membered heteroaryl (e.g., pyridyl or pyrimidyl). N-linked heterocyclic groups are linked via a component nitrogen atom. 4-to 7-membered heterocycloalkyl groups include, for example, piperidinyl, piperazinyl. pyrrolidinyi, azepanyl, morpholino, thiomorpholino and 1,1-dioxo- thiomorphoIin-4-yl. Representative aromatic heterocycles include azocinyl, pyridyl, pyrimidyl, imidazolyl and tetrazolyl. In certain embodiments, preferred heterocycles are 5- to 7-membered heterocycles having a single saturated, partially unsaturated or aromatic heterocyclic ring with 5 to 7 ring members, 1 or 2 ring members independently chosen from N, O and S, with remaining ring members being carbon.
Certain heterocycles are attached via an indicated linker group (e.g., (heterocycle)alkyl, (heterocycle)alkoxy and (heterocycle)alkylamino groups). In each case the heterocycle is covalently
bound to the indicated linker group, each of which carries the definition set forth above. For example, a (4- to 10-membered heterocycle)Co-C2alkyl is a heterocyclic group, which may be saturated, partially saturated or aromatic, that contains from 4 to 10 ring members and that is linked via a single covalent bond or a methylene or ethylene linker. As used herein, "heteroary!" indicates a monocyclic, bicyclic or tricyclic ring system that comprises at least one 5- or 6-membered heterocyclic aromatic ring that contains from 1 to 4 (preferably from 1 to 3) heteroatoms independently chosen from N, O and S, with remaining ring atoms being carbon. If the total number of S and O atoms in the heteroaryl group exceeds 1, these heteroatoms are not adjacent to one another. It is generally preferred that the total number of S and O atoms in the heteroaryl group is not more than 2; in certain embodiments, the total number of S and O atoms in the aromatic heterocycle is not more than 1. Examples of heteroaryl groups include, but are not limited to, oxazolyl, pyranyl, pyrazinyl, pyrazolopyrimidinyl, pyrazolyl, pyridizinyl, pyridyl, pyrimidinyl, pyrrolyl, quinolinyl, tetrazolyl, thiazolyi, thienylpyrazolyl, thiophenyl, triazolyl, benzo[<i]oxazolyl, benzofuranyl, benzothiazolyl, benzothiophenyl, beπzoxadiazolyl, dihydrobenzodioxynyl, furanyl, imidazolyl, indoiyl, and isoxazolyl.
A "heterocycloalkyl" group is a heterocycle as described above, which is fully or partially saturated. In certain embodiments preferred heterocycloalkyl groups are 5- to 7-membered heterocycloalkyi groups having a single saturated ring with 5 to 7 ring members. I or 2 ring members independently chosen from N, O and S, and remaining ring members being carbon. Certain such heterocycloalkyl groups are 5- or 6-membered. A "heterocyc]oalkyICo-CnaIkyl" is a heterocycloalkyi group linked via a single covalent bond or
 group, such as a C1- Cjaikylene group.
group, such as a C1- Cjaikylene group.
The term "(3- to 10-membered cycle)" refers to any carbocycie or heterocycle that has from
3- to 10 ring members. "(3- to 10-membered cycle)Co-C2alkoxy" is any such carbocycie that is linked via an oxygen atom (C0) or a Cs-C2aikoxy moiety. Certain (3- to 10-membered cycle)C0-
C^alkoxy groups include "(3- to 10-membered cycle)-O-" moieties, such as phenoxy and
(cycloalkyl)oxy groups (e.g., cyclopentyloxy).
A "substituent," as used herein, refers to a molecular moiety that is covalently bonded to an atom within a molecule of interest. For example, a ring substituent may be a moiety such as a halogen, alkyl group, haloalkyl group or other group discussed herein that is covalently bonded to an atom (preferably a carbon or nitrogen atom) that is a ring member. Substituents of aromatic groups are generally covalently bonded to a ring carbon atom. The term "substitution" refers to replacing a hydrogen atom in a molecular structure with a substituent, such that the valence on the designated atom is not exceeded, and such that a chemically stable compound (i.e., a compound that can be isolated, characterized, and tested for biological activity) results from the substitution.
Groups that are "optionally substituted" are unsubstituted or are substituted by other than hydrogen at one or more available positions, typically I5 2, 3, 4 or 5 positions, by one or more
suitable groups (which may be the same or different). Optional substitution is also indicated by the phrase "substituted with from 0 to X substituents," where X is the maximum number of possible substituents. Certain optionally substituted groups are substituted with from 0 to 2, 3 or 4 independently selected substituents (i.e., are unsubstituted or substituted with up to the recited maximum number of substituents).
The term "MCH receptor" refers to any naturally-occurring mammalian (e.g., human, monkey, or canine) MCH type 1 or type 2 receptor, as well as chimeric receptors in which one or more domains of a naturally-occurring MCHlR or MCH2R are replaced with a corresponding domain of a different G protein-coupled receptor, such that the ability of the chimeric receptor to bind MCH and mediate a dose-dependent release of intracellular calcium is not diminished. MCH receptors for use within the various assays and other methods described herein include, for example, recombinantly expressed human MCHlR (e.g., SEQ ΪD NO:6 of US Patent No. 7,078,484), recombinant^ expressed human MCH2R (e.g.. Genbank Accession No. Z86090; SEQ ID NO:29 of U.S. Patent Application Publication Number 2003/0148457), monkey MCHlR or MCH 2R (e.g., SEQ ID NO:2, 34 or 36 of U.S. Patent No. 7,078,474) or canine MCH2R (e.g., SEQ ID NO:39 of U.S. Patent No. 7,078,474). Chimeric MCH receptors that may be used as described herein include, for example, those disclosed in U.S. Patent No. 7,078,474 and U.S. Patent Application Publication Number 2003/0148457.
A "MCH receptor modulator," also referred to herein as a "modulator," is a compound that alters (increases or decreases) MCH receptor activation and/or MCH receptor-mediated signal transduction. MCH receptor modulators specifically provided herein are aminopiperidines and related compounds. In certain embodiments, a modulator may exhibit an EC50 or IC50 at MCH receptor that is less than 1 micromolar, 500 11M, 200 nM, 100 nM, 50 nM, 25 nM or 10 nM in a standard calcium mobilization assay (as described in Example 10, herein) and/or an agonist- stimulated GTP gamma35S binding assay (as described in Example 8, herein). A modulator may be a MCH receptor agonist or antagonist, although antagonists are preferred for certain purposes described herein.
A MCH receptor modulator binds with "high affinity" if the Kj at a MCH receptor is less than 1 micromolar, preferably less than 500 nanomolar, 100 nanomolar or 10 nanomolar. A modulator binds "specifically" to MCH receptor if it binds to a MCH receptor (total binding minus nonspecific binding) with a K; that is 10-fold, preferably 100-fold, and more preferably 1000-fold, less than the Kj measured for modulator binding to other G protein-coupled receptors. For example, a modulator may have a K1 of 500 nanomolar or less in an MCH receptor ligand binding assay and a
K, of at least 1 micromolar in a dopamine receptor ligand binding assay, such as the assay described in Example 8 (pages 111-1 12) of PCT International Publication Number WO 02/094799, which is hereby incorporated by reference. Representative assays for determining K, at MCH receptor are provided in Examples 6 and 8-10, herein.
A modulator is considered an "antagonist" if it detectably inhibits MCH binding to MCH receptor and/or MCH-mediated signal transduction (using, for example, the representative assay provided in Example 6 or Example 9); in general, such an antagonist has a IC50 value of less than 1 micromoiar, preferably less than 100 nanomolar, and more preferably less than 10 nanomolar within the assay provided in Example 6 and/or the assay provided in Example 9. MCH receptor antagonists include neutral antagonists and inverse agonists.
An "inverse agonist" is a compound that reduces the activity of MCH receptor below its basal activity level in the absence of added ligand. Inverse agonists may also inhibit the activity of MCH at MCH receptor, and/or may also inhibit binding of MCH to MCH receptor. The ability of a compound to inhibit the binding of MCH to MCH receptor may be measured by a binding assay, such as the binding assays given in Example 6 or Example 9. The basal activity of MCH receptor, as well as the reduction in MCH receptor activity due to the presence of antagonist, may be determined from a calcium mobilization assay, such as the assay of Example 10, or an agonist- stimulated GTP gamma33 S binding assay, such as the assay described in Example 8. A "neutral antagonist" of MCH receptor is a compound that inhibits the activity of MCH at
MCH receptor, but does not significantly change the basal activity of the receptor (e.g., within an assay as described in Example 8 or Example 10 performed in the absence of ligand, MCH receptor activity is reduced by no more than 10%, more preferably by no more than 5%, and even more preferably by no more than 2%; most preferably, there is no detectable reduction in activity). Neutral antagonists may also inhibit ligand binding to MCH receptor.
As used herein a "MCH receptor agonist" is a compound that elevates the activity of the receptor above the basal activity level of the receptor (i.e., enhances MCH receptor activation and/or MCH receptor-mediated signal transduction). MCH receptor agonist activity may be identified using the representative assays provided in Examples 8 and 10. In general, such an agonist has an EC50 value of less than 1 micromoiar, preferably less than 100 nanomolar, and more preferably less than 10 nanomolar within one or both of the assays provided in Examples 8 and 10.
A "therapeutically effective amount" (or dose) is an amount that, upon administration, is sufficient to provide a discernible patient benefit. For example, a therapeutically effective amount may reduce symptom severity or frequency, and/or may result in detectable weight loss. Alternatively, or in addition, a therapeutically effective amount may improve patient status or outcome and/or prevent or delay disease or symptom onset. A therapeutically effective amount or dose generally results in a concentration of compound in a body fluid (such as blood, plasma, serum,
CSF, synovial fluid, lymph, cellular interstitial fluid, tears or urine) that is sufficient to alter the binding of ligand to MCH receptor in vitro (using an assay provided in Example 6 or Example 9) and/or MCH-mediated signal transduction (using an assay provided in Example 8 or Example 10).
A "disease or disorder associated with MCH receptor activation," as used herein is any condition that is characterized by inappropriate stimulation of MCH receptor, regardless of the
amount of MCH present locally, and/or that is responsive to modulation of MCH receptor activity (i.e., the condition or a symptom thereof is alleviated by such modulation). Such conditions include, for example, metabolic disorders (such as diabetes), heart disease, stroke, eating disorders (such as obesity and bulimia nervosa) and sexual disorders such as anorgasmic and psychogenic impotence, as well as other diseases and disorders recited herein.
A "patient" is any individual treated with an aminopiperidine or related compound as provided herein. Patients include humans, as well as other animals such as companion animals (e.g., dogs and cats) and livestock. Patients may be experiencing one or more symptoms of a condition responsive to MCH receptor modulation, or may be free of such symptom(s) (i.e., treatment may be prophylactic).
AMINOPIPERIDINES AND RELATED COMPOUNDS
As noted above, the present invention provides aminopiperidines and related compounds of Formula I. Within certain aspects, such compounds are MCH receptor modulators, and may be specific for a particular MCH receptor (e.g., type 1 or type 2) or may inhibit or enhance ligand binding to multiple MCH receptors. MCH receptor modulators may be used to modulate MCH receptor activity in vivo, especially in the treatment of metabolic, feeding and sexual disorders in humans, domesticated companion animals and livestock animals. Modulators may also be used within a variety of in vitro assays, such as assays for receptor activity, as probes for detection and localization of MCH receptors and as standards in assays of MCH binding and MCH-mediated signal transduction. In general, the MCH receptor modulators provided herein detectably modulate
MCH receptor activity at submicromoiar concentrations, preferably at subnanomolar concentrations.
Certain aminopiperidines and related compounds further satisfy Formula II, III, IV, V, VI,
VII, VIlI or IX as described above. Within Formulas II, III and IV, certain compounds further satisfy one or more of Formulas Ila-IIg, HIa-IIId and 3Va-IVc, respectively:

Formula Ha Formula Hb

Formula lie Formula Hd

Formula lie Formula Iff

Formula Hg

Formula HIa Formula lϊϊb

Formula I lie Formula HId

Formula IVa Formufa IVb

Formula IVc
wherein p is 0. 1, 2 or 3 and s is 2 or 3.
Within the each of the Formulas provided herein, variables are generally as described above, fπ certain embodiments of the Formulas provided herein, one or more of the variables are as follows.
The R1 Variable
Within certain aminopiperidines and related compounds of any of the Formulas provided herein, R] groups satisfy the formula -L-M, in which L and M are as noted above. It will be apparent that the "L" moiety in such groups is divalent and is directly finked both to M and to the aromatic ring that comprises U and T. If L is a single covalent bond, then the "M" moiety is linked via a single covalent bond to the aromatic ring.
Within certain aminopiperidines and related compounds provided herein, Ri is a group of the Formula Rs-O-, in which linkage is via the oxygen atom and Rg is as defined for Formula IL Within certain such compounds, R8 is hydrogen, Cj-Qalkyl, C2-C6alkenyl, C2-C6alkynyl, mono- or di-(Ci-C3alkyl)aminoCi-C6alkyi or (5- to 7-membered heterocycle)C0-C6alkyl. each of which is substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosulfonyl, C]-C4alkyl, Ci-C^alkoxy and mono- and di-(Cr Csalkyl)aminoC(rC6alkyl. Certain R, groups are Ci-C6alkoxy, such as methoxy; other R| groups are substituted CrC6alkoxy, as described above.
The Variables P, Q, U and T Within certain aminopiperidines and related compounds provided herein, Q is substituted carbon. Within other such compounds, P is CR55, and at least one of P and Q is substituted carbon. In certain embodiments. R9 and Ri5 are independently hydrogen, CrC2alkyl, CrC2alkoxy or CV Cihaloalkyl; within further embodiments, R9 and RS 5 are independently CrC2aikyl, C]-C2alkoxy or Cj-Cihaloalkyi. Rg and Rj5 are both methyl within other embodiments. The variables U and T. within certain compounds provided herein, are both CR7. In certain embodiments, each R7 is hydrogen, C]-C2alkyL CrC2aIkoxy or Ci-C2haloalkyl; in further embodiments, each R7 is hydrogen. Within certain embodiments, R9 and Rj 5 are independently chosen from Ci-C4alkyl and each R7 is independently hydrogen, CrC2alkyl, CrC2alkoxy or Q- Cjhaloalkyl.
R3
Within certain aminopiperidines and related compounds. R3 is hydrogen. Within other such compounds, R3 is not H (e.g., R3 is methyl or ethyl). Within certain such compounds, the group

The variables m and n represent integers, as indicated above. In certain aminopiperidines and related compounds, n is 0, 1 or 2. Representative combinations for n and m include, for example, (a) n is 0 and m is 1, 2 or 3; (b) n is 1 and m is 2 or 3; (c) n is 2 and m is 0, 1 or 2; and (d) n is 3 and m is 0 or 1 . Certain n and m combinations are illustrated in the following table, in which the n and m values are given along with the resulting core ring:



Such core rings may, but need not, be substituted as provided herein (e.g., by the formation of a bridge).
Within certain aminopiperidines and related compounds of the Formulas provided herein, R2 represents zero substituents. Within other compounds, R? represents from 0 to 4 substituents independently chosen from Cj-dalkyl and
 Within still further compounds provided herein, two R2 groups are taken together to form a bridge. Within representative bridged
Within still further compounds provided herein, two R2 groups are taken together to form a bridge. Within representative bridged
compounds, the group designated

The variable q, where present, is 0 in certain embodiments and 1 in other embodiments. Representative R10 moieties include, for example, methyl, ethyl, propyl and butyl.
The X variable
Within certain aminopiperidines and related compounds of the Formulas provided herein, X is C(O). Within other compounds, X is C(O)-[C(R, iRi2)]v (e.g., C(O)CH2 or C(O)CH2CH2), C(O)N(Rn) {e.g., C(=O)NH) or C(=O)-[C(R,,R,2)]VY (e.g., C(O)CH2O or C(O)CH2CH2O). In each of these X moieties, it will be apparent that the orientation of the X group is retained (i.e., the carbonyl of each of the foregoing groups is linked to the nitrogen atom adjacent to the X moiety in the Formula via a single covalent bond).
As noted above, Rn and R!2 are independently H, Ci-Cgalkyl, CrQalkoxy, C2-C<;alky1 ether, or taken together with a RB or R6 moiety to form a Cs-Cycycioalkyl or heterocycloalkyl. It will be apparent that, if RA is a cyclic moiety, such a cycloalkyl or heterocycloalkyl will be a ring that is fused to RA. If X is C(O)NRn, Rn is preferably H, C,-C6alkyl or C2-C6alkyl ether.
The RA variable
RA, within certain aminopiperidines and related compounds of the Formulas provided herein, is (e.g., methyl, ethyl, propyl, butyl or pentyl);
 (e.g., trifluoroethyl);
(e.g., trifluoroethyl);
 (e.g., cyclopropyl, cycϊobutyl, cyclopentyl. cyclohexyl or cycloheptyl, each of which is linked via a single covalent bond or a methylene or ethylene linker); or (5- to 7-membered heterocycloalkylJCo-C^alkyl (e.g., tetrahydrofuranyl, piperidinyl or piperazinyl; each of which is linked via a single covalent bond or a methylene or ethylene linker), each of which is optionally substituted with one or more RB as described above. RA is (Cj- C7cycloalkyi)Co-C2alkyl within certain such compounds. Representative such compounds of
(e.g., cyclopropyl, cycϊobutyl, cyclopentyl. cyclohexyl or cycloheptyl, each of which is linked via a single covalent bond or a methylene or ethylene linker); or (5- to 7-membered heterocycloalkylJCo-C^alkyl (e.g., tetrahydrofuranyl, piperidinyl or piperazinyl; each of which is linked via a single covalent bond or a methylene or ethylene linker), each of which is optionally substituted with one or more RB as described above. RA is (Cj- C7cycloalkyi)Co-C2alkyl within certain such compounds. Representative such compounds of
Formula II further satisfy Formula Hh:
Formula Hh
 wherein f is O, 1, 2 or 3. Certain compounds of Formula Hh further satisfy one of Formulas IIh-1 to Hh-3:
wherein f is O, 1, 2 or 3. Certain compounds of Formula Hh further satisfy one of Formulas IIh-1 to Hh-3:

Formula Hh- 1 Formula IIh-2

Formula IIh-3 wherein n is 0. 1 or 2.
Other compounds of Formula II further satisfy Formula Hi:
Formula Hi
 wherein f is 0, 1, 2 or 3. Still further compounds of Formula Hi further satisfy one of Formulas Hi-I to IIi-3:
wherein f is 0, 1, 2 or 3. Still further compounds of Formula Hi further satisfy one of Formulas Hi-I to IIi-3:


Formula Iϊi-3 wherein n is 0. I or 2.
Other representative compounds of Formula II further satisfy Formula ITj or Formula Hk:


Formula llk-3 wherein n is 0, 1 or 2.
Within certain embodiments, each RB is independently (i) halogen, hydroxy, nitro or cyano; or (ii) C,-C6alkyl, C2-Qalkenyl, C2-C6alkynyl, Ci-C6alkanoyl, C]-C6alkyltliio, C]-C6alkanoyloxy, Ci-Qalkoxycarbonyl, C|-C6alkylsulfonyl, Q-Qhaloafkyl, (C1-C7cycloalkyI)C0-C2alkyl, (4- to 7- membered heteiOcycle)Co-C2alkyl or phenylC0-C2alky!; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Ci-C4alky], Ci - C4alkoxy, C]~C4haloalky!, CrC}haloalkoxy, mono- or di-(CrC6alkyl)amino and 5- or 6-membered heterocycloalkyl.
Within other aminopiperidines and related compounds of the Formulas provided herein, RA is 6- to 10-membered aryl {e.g., phenyl) or 5- to 10-membered heteroaiyl (e.g., a 5- or 6-membered heteroaryl such as pyridyl, pyrimidyl, thiazolyl, thiophenyl, imidazolyl, triazolyL tetrazolyl or furanyl; or a 9- or 10-membered heteroaiyl such as indolyl, IH-indolyl, isoindolyl, benzofuranyl, dihydrobenzofuranyl. benzisoxazolyl, benzothiadiazolyl, benzoxadiazolyl. benzotliiophenyi, benzodioxolyl, dihydrobenzodioxinyl, quinolinyl, quinazolinyl, and the like), each of which is
optionally substituted as described above. In certain such compounds, RA is H E~~D' ; wherein A and E are independently M or CR5; B, J and D are independently N or CR6; each R5 is independently: (i) hydrogen, halogen, nitro, cyano or -COOH; (ii) Cj-C6alkyl, C2-C6alkenyl, C2- C6alkynyl, Ci-C6alkanoyl, C3-C6a!kanone, C2-C6alkyl ether, CrC6alkoxycarbonyl, C1- C6alkylsulfonyl or Ci-C6haloalkyl; each of which is substituted with from 0 to 3 substituents independently chosen from hydroxy, amino, cyano, Ci-C6alkoxy and mono- or di-(Cr C6alkyl)amino; or (iii) taken together with a Rn or R12 moiety to form a C5-C7cycloalkyl or a 5- to 7-membered heterocycloalkyl; and each R6 is independently: (i) hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl; or (ii) Ci-C6alkyl, C2-Qaikenyl, C3- C6alkynyl, CrC6aIkoxy, CrC6alkanoyl, Ci -Chalky ithio, CrC6alkanoyloxy, Ci-C6alkoxycarbonyl, CrC6alky!sulfonyl, C,-C6haloalkyl, mono- or di-(CrC6alkyi)ammoCo-C4a!kyl, mono- or di-(Cr
C6aIkyl)aminocarbonylCo-C4a!kyi, mono- or di-(Ci-C6alkyl)aminosuIfonylCo-Cjalkyl, (C3- C7cycloalkyI)Co-C2alky!, (4- to IO-membered heterocycle)C0-C2aIkyL phenylC0-C2alkyl or (3- to 10-membered cycle)Co-C2alkoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C!~C4alkyl, CpGialkoxy, Cr C4haloalkyl,
 mono- or di-(Ci-C6alkyl)amino, or 5- or 6-membered heterocycloalkyi ; or two adjacent R6 groups are taken together to form a 5- or 6-membered ring that is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CpGtalkyl., CpC4alkoxy,
mono- or di-(Ci-C6alkyl)amino, or 5- or 6-membered heterocycloalkyi ; or two adjacent R6 groups are taken together to form a 5- or 6-membered ring that is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CpGtalkyl., CpC4alkoxy,
 and mono- or di-(Ci-C6alkyl)amino.
and mono- or di-(Ci-C6alkyl)amino.
Within certain such compounds, each R5 is independently: (i) hydrogen, halogen, nitro, cyano or -COOH; or (ii) CpC6alkyl, C2-C6alkenyl, C2-C<;aikynyl, CpQalkanoyl, Cs-C^alkanone,
C2-C6alkyl ether. CpCealkoxycarbonyϊ, CpC6aIkylsulfony! or CpQhaloalkyl; each of which is substituted with from O to 3 substituents independently chosen from hydroxy, amino, cyano, Cp
Cgalkoxy and mono- or di-(CpC6alkyI)amino; and each R6 is independently: (i) hydrogen, halogen, hydroxy, nitro, cyano or-COOH; or (ii) CpQalkyl, C2-Qalkenyl, C2-Qalkynyl, CpCgalkoxy, C]- Cfialkanoyl, Cj-Cβalkylthio, CpC6aIkanoyIoxy, Ci-C6alkoxycai'bonyl, CpC6alkyIsulfonyl, C]-
Cfihaloalkyl, (Cs-CvCycloalkyOCo-Cialkyl, (4- to 7-membered heterocycle)C0-C2alkyl or pheny!C0-
C2alkyl; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CpC4aIkyl, CrC4alkoxy, CpQhaloalkyl, CrC4haloalkoxy, mono- or di-(CrC6alkyl)ammo and 5- or 6-membered heterocycloalkyi; or two adjacent R6 groups are taken together to form a fused, 5- or 6-membered ring. Representative such compounds of
Formula II further satisfy Formula 11-1:
Formula II— 1
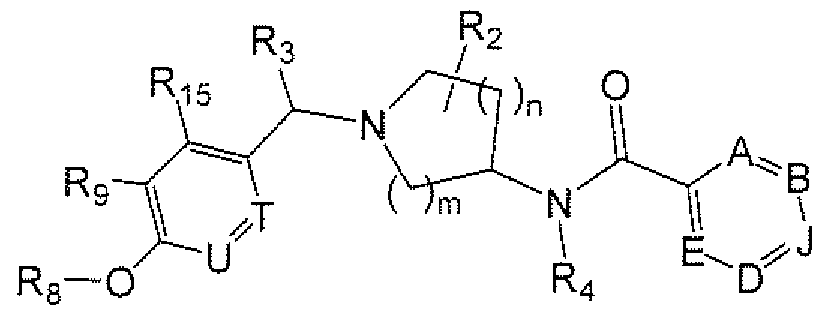
Certain compounds of Formula IT-I further satisfy one of Formulas II- 1-1 to 11-1-3:

Formula ϊϊ-1-2

Formula II-1-3 wherein n is O, 1 or 2.
Other compounds of Formula II further satisfy Formula Hm:
Formula llm

Still further compounds of Foiτnuia ϊlm further satisfy one of Formulas 1Im-I to lϊm-3:


Formula IIm-3 wherein n is O, 1 or 2.
Other representative compounds of Formula Il further satisfy Foπnula On or Formula Ho:
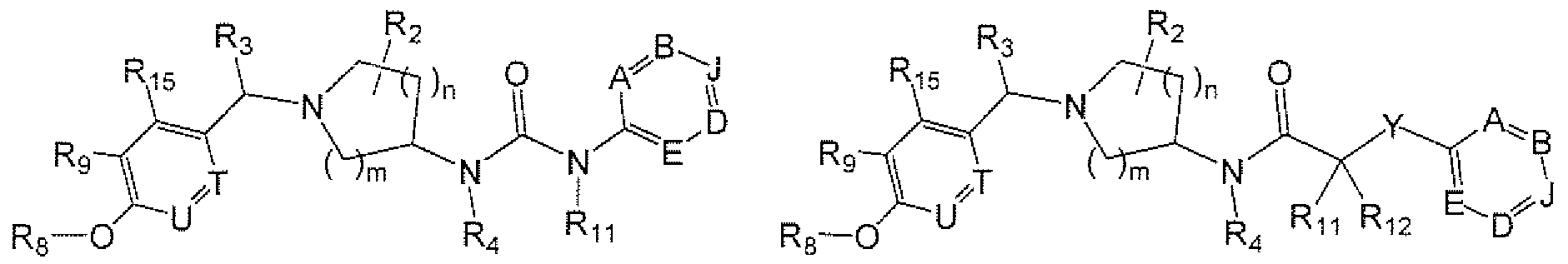
Formula Hn Formula Ho
Representative compounds of Formula Ho further satisfy one of Formulas Ho-I to IIo-3:

Foπnula Ho- 1 Formula IIo-2

Formula Ho-3 wherein n is O, 1 or 2.
It will be apparent that analogous Formulas, specifying X, R4 and/or RA moieties as illustrated above, may be generated for Formulas III-IX. Such analogous Formulas are encompassed by the present invention.
Within certain aminopiperidines and related compounds of the Formulas provided herein, at least one of B, J and D is substituted carbon. For example, in certain such compounds, J is substituted carbon, B is substituted carbon, or both J and B are substituted carbon. In other such compounds, at least two, or exactly two, of B, J and D are carbon that is substituted (e.g., with a substituent independently chosen from methyl, ethyl, halogen, C]-C2HaIOaIkVl and C]-C2alkoxy).
Within further embodiments. A, J and E are optionally substituted carbon and D is N; in certain such compounds A and E are each CH.
Within further aminopiperidines and related compounds of the Formulas provided herein,
RA is E-G ; E-A or D-E ; wherein A, B, E and D are independently N or CR6; G is NR6, S or O; and R6 is as described above. Representative such RA groups include, for
example, R6

R5 and R6 are generally as indicated above. In certain embodiments of the above Formulas, each R6 is independently: (i) hydrogen, halogen, hydroxy, nitro or cyano; or (ii) d-Qalkyl, C2- Qalkenyl, C2-C6alkynyl, CrC6alkoxy, CrC6alkanoyl, CrC6alkylthio, CrQalkanoyIoxy, C1- C6alkoxycarbony], C]-C6alky]sulfonyl, C,-C6haloaikyl, (C3-C7cycioalkyl)Co-C2aikyl, (4- to 7- membered heteiOcycle)C,-C2alkyl or phenyiCo-C2alkyl; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Ci-C4alkyl, C1- C.talkoxy, CrC4haloalkyi, Ci-C4haloalkoxy, mono- or di-(CrC6alkyl)aniino and 5- or 6-membered heterocycloalkyl. In other embodiments, one substituent represented by R3 or R6 is taken together with Rn or R]2 to form a cycloalkyl or heterocycloalkyl ring. It will be apparent that, if RA is a cyclic moiety, the cycloalkyi or heterocycloalkyl ring so formed will result in fused rings (e.g., benzodioxolyl). In further embodiments, two substituents represented by R6 are taken together to foπn a carbocycle or heterocycle.
In certain compounds of Formula VIl, VIII or IX, RA is 6- to 10-membered aryl or 5- to 10- membered heteroaryl, each of which is substituted with from 0 to 4 substituents independently
chosen from RB. Representative RA groups include D . wherein A and E are independently
N or CR5: B, J and D are independently N or CR^; and each R5 and R5 is independently: (i) hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl; (ii) Cr C6alkyl, C2-C6alkenyl, C2-C6aikynyl, C,-C6alkoxy, CrC6alkanoyl, CrC6alkylthio, C1- Cfialkanoyloxy, C rC6alkoxycarbonyl; CpC6alkylsulfonyl, CpCβhaloalkyl, mono- or di-(Cr C6alkyl)aminoC0-C4alkyl, mono- or di-(CpC6alkyl)aminocarbonylCo-C4alkyl, mono- or di-(C,- C6alkyl)aminosulfonylCo-C4alkyl, (C3-C7cycloalkyl)C0-C2alkyi, (4- to 10-membered heterocycie)C0- C2alkyl, phenyIC0-C2alkyl or (3- to 10-membered cyc]e)C0-C2aikoxy; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Cp C4alkyl, CpC4alkoxy, C]-C4haloalkyi, CpC4haIoalkoxy, mono- or di-(CpC6alkyi)amino, or 5- or 6- membered heterocycioalkyl. Representative R5 groups include (i) hydrogen, halogen, nitro, cyano or -COOH; and (ii) C,-C6alkyi, CrC6alkenyl, C2-C6alkynyl, CpQalkanoyL C3-C6aikanone, C2- C6alkyl ether, CpC6alkoxycarbonyl, CpCealkylsulfonyl or CpCshaloalkyl; each of which is substituted with from O to 3 substituents independently chosen from hydroxy, amino, cyano, Cj- Qaikoxy and mono- or di-(CrC6alky!)amino; and representative R/, groups include: (i) hydrogen, halogen, hydroxy, nitro, cyano or-COOH; and (ii) CpQalkyl, Q-Qalkenyl, Ci-Cβalkynyl, Cr C6alkoxy, CrC6alkanoyl, C]-C6alkylthio, C]-C6alkanoyloxy, C]-C6alkoxycarbonyl, Cj- Qalkyisulfoπyl, Cj-Cήhaloalkyl, (C3-C7cycloalkyl)Co-C2alkyl, (4- to 10-membered heterocycle)C0- C2alkyl or phenylC0-C2alkyl; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Ci-C4alkyl, C)-C4alkoxy, Cj- C4ha!oalkyl, C]-C4haloalkoxy, mono- or di-(C|-Cbalkyl)amino and 5- or 6-membered heterocycioalkyl; or two adjacent R5 groups are taken together to form a fused, 5- or 6-membered ring, In certain such compounds, at least one of B, J and D is substituted carbon (e.g., J or B is substituted carbon or two of B, J and D are substituted carbon). Other RA groups include Q-Cgalkyl, Cs-C^haloalkyl, (C3-C3cycIoalkyl)C0-C4alkyl or (5- to
7-membered heterocycloalkyOCo-Cjalkyl, each of which is substituted with from 0 to 4 substituents independently chosen from: (i) halogen, hydroxy, nitro or cyano; and (ii) CpCβalkyl, C2-C6alkenyl, CrQalkynyl, CrQalkanoyl, Ci-C6alkylthio, Ci-Csalkanoyloxy, Ci-Cealkoxycarbonyl, C3- C5alkylsulfonyi, C1-Ct>haloalkyl, (C3-C7cycloalkyl)C0-C2alkyl, (4- to 7-membered heterocycle)C0- C2alkyl or phenylCo-C2alkyl; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Ci-C4alkyl, Ci-C4alkoxy, Ci- C4haloalkyl, CpC4HaIOaIkOXy, mono- or di-(CpCgalkyl)amino and 5- or 6-membered heterocycioalkyl.
Still further RA groups are 5-membered heteroaryl groups that are substituted with from 0 to 3 substituents independently chosen from: (i) halogen, hydroxy, nitro, cyano, amino, -COOH,
aminocarbonyl or aminosulfonyl; (ii) Ci-C6aϊkyl, CrQaSkenyl, C2-C6alkynyl, Cj-Cβalkoxy, C,- Cealkanoyl, CrCgalkylthio, Cj-C<salkanoyloxy, C]-C6alkoxycarbonyi, Ci-Cgalkylsulfoπyl, Q- Cfehaioalkyl, mono- or di-(CrC6aIkyl)ammoC0-C4aIkyl, mono- or di-(Ci-C6aIky!)aminocarbonylCo- C4alkyl, mono- or di-(Cl-C6a]kyl)aminosulfonyIC0~C4alkyl, (C3-C7cycloalky!)Co-C2aIky], (4- to 10- membered heterocycle)C0-C2alkyl, phenylC0-C2alkyl or (3- to 10-membered cycle)Co-C2alkoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Ci-C4alkyl, Q-C4alkoxy,
 Ci-C4haloalkoxy, mono- or di-(CrC6alky!)amino, or 5- or 6-membered heterocycloalkyl.
Ci-C4haloalkoxy, mono- or di-(CrC6alky!)amino, or 5- or 6-membered heterocycloalkyl.
Within certain compounds of Formula ViI, VIIl or IX3 the variable X is C(=O). Within certain compounds of Formula VII, VIIl or IX, at least one of R9 and R15 is not H
(e.g., Rg and R! 5 are both other than hydrogen; R? and R!5 are independently C|-C2alkyl, C]- C2aikoxy or C)-C2haloalkyl; R9 and R,5 are each methyl; or R9 and R|5 are independently chosen from C;-C2alkyl and each R7 is independently hydrogen, Cj-C2alkyl, C|-C2alkoxy or Q^haloalkyl. The variable R^ in certain compounds of Formula VII, VIII or IX, is not H. In certain such compounds, Rj is OR8; and R8 is hydrogen, Ci-C8alkyl, C2-C3alkenyl. C2-C8alkynyl, C2-C3alkyl ether, mono- or di-(Ci-C3alkyl)aminoCo-C6alkyI, (C3-C7cycloalky!)Co-C6alkyl or (5- to 10- membered heterocycle)C0-C6aIkyl, each of which is substituted with from O to 3 substituents independentiy chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosulfony], -COOH, CrC6alkyl, Ci-C6alkoxy, d-Qhaloalkyl, CrC6haloalkoxy, mono- or di- (Ci-C6a!kyl)aminoC0-C6alkyl, CrC6alkylsulfonyl, CrC6aIky]thio, CrC6alkyIaminosulfonyl, C1- C6alkoxycarbonyl or C] -Cc,alkanoyl amino. In further such compounds, R8 is hydrogen, CrC6alky1, C2-C(,alkenyl, C2-C6alkyny], mono- or di-(CrC8alkyl)aminoC]-C6alkyl or (5- to 7-membered heterocycle)Co-C<jalkyl, each of which is substituted with from O to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosuifonyl, C1- C4alkyi,
 and mono- and di-(C!-C8alkyl)aminoCo-C6a]kyl. In further such compounds, Ri is methoxy.
and mono- and di-(C!-C8alkyl)aminoCo-C6a]kyl. In further such compounds, Ri is methoxy.
Representative aminopiperidines and related compounds provided herein include, but are not limited to, those specifically described in Examples 1-4, herein. It will be apparent that the compounds recited therein are representative only, and are not intended to limit the scope of the present invention. Further, as noted above, all compounds may be present as a free base, a pharmaceutically acceptable salt (such as an acid addition salt) or other form, such as a solvate (e.g., hydrate).
In certain embodiments, aminopiperidines and related compounds provided herein detectably alter (modulate) MCH binding to MCHlR and/or MCH2R, as determined using a standard in vitro MCH receptor ligand binding assay and/or functional assay. References herein to a
11MCH receptor ligand binding assay" refer to either of the assays provided in Examples 6 and 9.
Within such assays, the receptor is incubated with labeled MCH (or other suitable ligand) and a test
compound. A test compound that detectably modulates binding of ligand to MCH receptor will result in a decrease or increase in the amount of label bound to the MCH receptor preparation, relative to the amount of label bound in the absence of the compound. Preferably, such a compound will exhibit a K1 at an MCH receptor that is less than 1 micromolar, more preferably less than 500 nM, 100 nM, 20 nM or 10 nM, within an assay performed as described in Example 6 and/or within an assay performed as described in Example 9. Certain preferred compounds are MCH receptor antagonists, and exhibit IC50 values of about 4 micromolar or less, more preferably 1 micromolar or less, still more preferably about 100 nanomolar or less, or 10 nanomolar or less within a standard in vitro MCH receptor mediated calcium mobilization assay, as provided in Example 10 and/or an agonist-stimulated GTP gamma35S binding assay, as described in Example 8.
If desired, aminopiperidines and related compounds provided herein may be evaluated for certain pharmacological properties including, but not limited to, oral bioavailability (preferred compounds are orally bioavailable to an extent allowing for oral doses of less than 140 mg/kg, preferably less than 50 mg/kg, more preferably less than 30 mg/kg, even more preferably less than 10 mg/kg, still more preferably less than 1 mg/kg), toxicity (a preferred compound is nontoxic when a therapeutically effective amount is administered to a subject), side effects (a preferred compound produces side effects comparable to placebo when a therapeutically effective amount of the compound is administered to a subject), serum protein binding and in vitro and in vivo half-life (a preferred compound exhibits an in vitro half-life that is equal to an in vivo half-life allowing for Q. LD. dosing, preferably T.I.D. dosing, more preferably B. I. D. dosing, and most preferably once-a- day dosing). In addition, differential penetration of the blood brain barrier may be desirable for compounds used to treat CNS disorders, while low brain levels of compounds used to treat peripheral disorders are preferred. Routine assays that are well known in the art may be used to assess these properties and identify superior compounds for a particular use. For example, assays used to predict bioavailability include transport across human intestinal cell monolayers, including Caco~2 cell monolayers. Penetration of the blood brain barrier of a compound in humans may be predicted from the brain levels of the compound in laboratory animals given the compound (e.g., intravenously). Serum protein binding may be predicted from albumin binding assays. Compound half-life is inversely proportional to the frequency of dosage of a compound. In vitro half-lives of compounds may be predicted from assays of microsomal half-life as described in Example 12.
As noted above, preferred aminopiperidines and related compounds provided herein are nontoxic. In general, the term "nontoxic" shall be understood in a relative sense and is intended to refer to any substance that has been approved by the United States Food and Drug Administration ("FDA") for administration to mammals (preferably humans) or, in keeping with established criteria, is susceptible to approval by the FDA for administration to mammals (preferably humans). In addition, a highly preferred nontoxic compound generally satisfies one or more of the following criteria when administered in minimum therapeutically effective amounts, or when contacted with
cells at a concentration that is sufficient to inhibit the binding of Iigand to MCH receptor in vitro: (1) does not substantially inhibit cellular ATP production; (2) does not significantly prolong heart QT intervals; (3) does not cause substantial liver enlargement and (4) does not cause substantial release of liver enzymes. As used herein, a compound that does not substantially inhibit cellular ATP production is a compound that satisfies the criteria set forth in Example 1 1. In other words, cells treated as described in Example 1 1 with 100 μM of such a compound exhibit ATP levels that are at least 50% of the ATP levels detected in untreated cells. In more highly preferred embodiments, such cells exhibit ATP levels that are at least 80% of the ATP levels detected in untreated cells. The concentration of compound used in such assays is generally at least 10-fold, 100-fold or 1000-fold greater than the EC50 or IC50 for the modulator in the assay of Example 8 or Example 10.
A compound that does not significantly prolong heart QT intervals is a compound that does not result in a statistically significant prolongation of heart QT intervals (as determined by electrocardiography) in guinea pigs, minipigs or dogs upon administration of a dose that yields a serum concentration equal to the ECjo or IC50 for the compound. In certain preferred embodiments, a dose of 0.01, 0.05, 0.1 , 0.5, 1, 5, 10, 40 or 50 mg/kg administered parenterally or orally does not result in a statistically significant prolongation of heart QT intervals. By "statistically significant" is meant results varying from control at the p<0.1 level or more preferably at the p<0.05 level of significance as measured using a standard parametric assay of statistical significance such as a student's T test.
A compound does not cause substantial liver enlargement if daily treatment of laboratory rodents (e.g., mice or rats) for 5-10 days with a dose that yields a serum concentration equal to the EC50 or IC50 for the compound results in an increase in liver to body weight ratio that is no more than 100% over matched controls. In more highly preferred embodiments, such doses do not cause liver enlargement of more than 75% or 50% over matched controls. If non-rodent mammals (e.g., dogs) are used, such doses should not result in an increase of liver to body weight ratio of more than 50%, preferably not more than 25%, and more preferably not more than 10% over matched untreated controls. Preferred doses within such assays include 0.01, 0.05. 0.1 , 0.5, 1, 5, 10. 40 or 50 mg/kg administered parenterally or orally. Similarly, a compound does not promote substantial release of liver enzymes if administration of twice the minimum dose that yields a serum concentration equal to the ECJO or IC50 for the compound does not elevate serum levels of ALT, LDH or AST in laboratory rodents by more than 100% over matched mock-treated controls. In more preferred embodiments, such doses do not elevate such serum levels by more than 75% or 50% over matched controls. Alternatively, a compound does not promote substantial release of liver enzymes if, in an in vitro hepatocyte assay, concentrations (in culture media or other such solutions that are contacted and incubated with hepatocytes in vitro) that are equal to the EC50 or IC50 for the compound do not cause detectable
release of any of such liver enzymes into culture medium above baseline levels seen in media from matched mock-treated control cells. In more highly preferred embodiments, there is no detectable release of any of such liver enzymes into culture medium above baseline levels when such compound concentrations are five-fold, and preferably ten-fold, the EC50 or IC5Q for the compound. In other embodiments, certain preferred compounds do not inhibit or induce microsomal cytochrome P450 enzyme activities, such as CYP 1A2 activity, CYP2A6 activity. CYP2C9 activity, CYP2C19 activity, CYP2D6 activity, CYP2E1 activity or CYP3A4 activity at a concentration equal to the EC50 or IC50 for the compound.
Certain preferred compounds are not clastogenic (e.g., as determined using a mouse erythrocyte precursor cell micronucieus assay, an Ames micronucleus assay, a spiral micronucleus assay or the like) at a concentration equal the EC50 or IC50 for the compound. In other embodiments, certain preferred compounds do not induce sister chromatid exchange (e.g., in Chinese hamster ovaiy cells) at such concentrations.
For detection purposes, as discussed in more detail below, aminopiperi dines and related compounds provided herein may be isotopically-iabeled or radiolabeled. For example, compounds of Formula I may have one or more atoms replaced by an atom of the same element having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes that can be present in the compounds provided herein include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2H, Η, 1 1C, 'JC, 14C. 15N, '8O, 17O, 31P, 32P, 35S, 18F and 36Cl. In addition, substitution with heavy isotopes such as deuterium (i.e., "H) can afford certain therapeutic advantages resulting from greater metabolic stability, such as increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some circumstances.
PHARMACEUTICAL COMPOSITIONS Aminopiperidines and related compounds can be administered as the neat chemical, but are preferably administered as a pharmaceutical composition comprising such a compound, together with at least one physiologically acceptable carrier or excipient. Representative carriers include, for example, water, buffers (e.g., neutral buffered saline or phosphate buffered saline), ethanof, mineral oil, vegetable oil, dimethyl sulfoxide, carbohydrates (e.g., glucose, mannose, sucrose or dextrans), mannitol and proteins. Additional optional components include adjuvants, diluents, polypeptides or amino acids such as glycine, antioxidants, chelating agents such as EDTA or glutathione and/or preservatives. Preferred pharmaceutical compositions are formulated for oral delivery to humans or other animals (e.g., companion animals such as dogs or cats). In addition, other active ingredients may (but need not) be included in the pharmaceutical compositions provided herein. Pharmaceutical compositions may also optionally comprise an activity enhancer, chosen from a wide variety of molecules that function in different ways to enhance MCH receptor modulator effect. Particular classes of activity enhancers include skin penetration enhancers and absorption enhancers,
Pharmaceutical carriers must be of sufficiently high purity and sufficiently low toxicity to render them suitable for administration to the animal being treated. The carrier can be inert or it can possess pharmaceutical benefits. The amount of carrier employed in conjunction with the compound is sufficient Io provide a practical quantity of material for administration per unit dose of the compound. Representative pharmaceutically acceptable carriers or components thereof are sugars, such as lactose, giucose and sucrose; starches, such as corn starch and potato starch; cellulose and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and methyl cellulose; powdered tragacanth; malt; gelatin: talc; solid lubricants, such as stearic acid and magnesium stearate; calcium sulfate; synthetic oils; vegetable oils, such as peanut oil, cottonseed oil, sesame oil, olive oil and corn oil; polyols such as propylene glycol, glycerine, sorbitol, mannitol and polyethylene glycol; alginic acid; phosphate buffer solutions; emuisifiers, such as the TWEENS; wetting agents, such as sodium lauryi sulfate; coloring agents; flavoring agents; tableting agents; stabilizers; antioxidants; preservatives; pyrogen-free water; isotonic saline; and phosphate buffer solutions. To prepare a pharmaceutical composition, effective concentrations of one or more aminopiperidines or related compounds provided herein are mixed with one or more suitable pharmaceutical earners or excipients. In instances in which the compounds exhibit insufficient solubility, methods for solubilizing compounds may be used. Such methods are known to those of skill in this art and include, but are not limited to, using cosolvents such as dimethylsulfoxide (DMSO), using a surfactant, such as TWEEN, or dissolution in aqueous sodium bicarbonate. Upon mixing or addition of the compound(s), the resulting mixture may be a solution, suspension, emulsion or the like. The form of the resulting mixture depends upon a number of factors, including the intended mode of administration and the solubility of the compound in the chosen carrier.
Pharmaceutical compositions may be formulated for administration by any suitable route, including orally, topically, parenterally, by inhalation (e.g., nasal or oral) or spray, sublingually, transdermally, via buccal administration, rectal Iy, as an ophthalmic solution or by other means, and may be prepared in dosage unit formulations and/or formulated as a lyophilizate. The term parenteral as used herein includes subcutaneous, intradermal, intravascular (e.g., intravenous), intramuscular, spinal, intracranial, intrathecal and intraperitoneal injection, as well as any similar injection or infusion technique.
Dosage formulations suitable for oral use include, for example, tablets, troches, lozenges, liquid solutions, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, tinctures, syrups or elixirs. Typical components of carriers for syrups, elixirs, emulsions and suspensions include ethanol, glycerol, propylene glycol, polyethylene glycol, liquid sucrose, sorbitol and water. Such formulations may also contain a demulcent. Alternatively, formulations containing these compounds can be presented as a dry product (optionally as an
admixture with a dispersing or wetting agent, suspending agent and one or more preservatives) for constitution with water or other suitable vehicle before use.
Aqueous suspensions comprise the active materials ) in admixture with one or more suitable excipients, such as suspending agents (e.g., sodium carboxymethylcellulose, methylcellulose, hydroxypropyjmethylcellulose, AVICEL RC-591, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia); and dispersing or wetting agents (e.g., polysorbate 80, naturaily- occurring phosphatides such as lecithin, condensation products of an alkylene oxide with fatty acids such as polyoxyethylene stearate, condensation products of ethylene oxide with long chain aliphatic alcohols such as heptadecaethyleneoxycetanol, condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate. or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides such as polyethylene sorbitan monooleate).
Oily suspensions may be formulated by suspending the active ingredients in a vegetable oil (e.g., peanut oil, olive oil, sesame oil or coconut oil), a mineral oil (such as liquid paraffin) or a mixture of such oils. The oily suspensions may further contain a thickening agent, such as beeswax, hard paraffin or cetyl alcohol.
Pharmaceutical compositions provided herein may also be in the form of oil-in-water emulsions. The oily phase may be a vegetable oil, mineral oil, or mixture thereof as described above. Suitable emulsifying agents include naturally-occurring gums (e.g., gum acacia or gum tragacanth), naturally-occurring phosphatides (e.g., soy bean phosphatide, lecithin and esters or partial esters derived from fatty acids and hexitol), and anhydrides (e.g., sorbitan monoleate and condensation products of the above partial esters with ethylene oxide, such as polyoxyethylene sorbitan monoleate).
Excipients suitable for the manufacture of tablets and capsules include, for example, inert diluents to increase the bulk weight of the material to be tableted (e.g., calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate), granulating and disintegrating agents that modify the disintegration rate in the environment of use (e.g., corn starch, starch derivatives, alginic acid and salts of carboxymethylcellulose), binding agents that impart cohesive qualities to the powdered material(s) (e.g., starch, gelatin, acacia and sugars such as sucrose, glucose, dextrose and lactose) and lubricating agents (e.g., magnesium stearate, calcium stearate, stearic acid or talc).
GIidants such as silicon dioxide can be used to improve flow characteristics of the powder mixture.
Tablets may be formed using standard techniques, including dry granulation, direct compression and wet granulation. Tablets may be uncoated or may be coated by known techniques. Capsules include, for example, hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent (e.g., calcium carbonate, calcium phosphate or kaolin), as well as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium (e.g., peanut oil, liquid paraffin or olive oil).
Compositions intended for oral use may further contain one or more optional agents, such as sweetening agents (e.g., glycerol, propylene glycol, sorbitol, sucrose, aspartame, saccharin, menthol, peppermint or fruit flavor); suspending agents (e.g., sorbitol syrup, methyl cellulose, glucose/sugar, syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminum stearate gel and hydrogenated edible fats); emulsifying agents (e.g., lecithin, sorbitan monsoleate or acacia); nonaqueous vehicles such as edible oils (e.g., almond oil, fractionated coconut oil, silyl esters, propylene glycol and ethyl alcohol); preservatives (e.g., methyl or propyl p-hydroxybenzoate, sodium benzoate, methyl paraben, ascorbic acid and/or sorbic acid); flavoring agents; and/or coloring agents (e.g., the FD&C dyes), in order to provide pharmaceutically appealing and palatable preparations.
A pharmaceutical composition may be prepared as a sterile injectable aqueous or oleaginous suspension. The active ingredient(s), depending on the vehicle and concentration used, can either be suspended or dissolved in the vehicle. Such a composition may be formulated according to the known art using suitable dispersing, wetting agents and/or suspending agents such as those mentioned above. Among the acceptable vehicles and solvents that may be employed are water,
1,3-butanedioI, Ringer's solution and isotonic sodium chloride solution. In addition, sterile, fixed oils may be employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed, including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid find use in the preparation of injectable compositions, and adjuvants such as local anesthetics, preservatives and/or buffering agents can be dissolved in the vehicle.
Pharmaceutical compositions may also be prepared in the form of suppositories (e.g., for rectal administration). Such compositions can be prepared by mixing the drug with a suitable non- irritating excipient that is solid at ordinary temperatures but liquid at the body temperature and will therefore melt in the body to release the drug. Suitable excipients include, for example, cocoa butter and polyethylene glycols.
Compositions for inhalation typically can be provided in the form of a solution, suspension or emulsion that can be administered as a dry powder or in the form of an aerosol using a conventional propel! ant (e.g., dichlorodifiuoromethane or trichiorofluoromethane).
Pharmaceutical compositions may be formulated for local or topical application, such as for topical application to the skin or mucous membranes. Topical compositions may be in any suitable form including, for example, solutions, creams, ointments, gels, lotions, milks, cleansers, moisturizers, sprays, skin patches and the like. Such solutions may, for example, be formulated as 0.01 %- 10% isotonic solutions, pH about 5-7, with appropriate salts. Pharmaceutical compositions may also be formulated for transdermal administration as a transdermal patch. Pharmaceutical compositions may be formulated for release at a pre-determined rate.
Instantaneous release may be achieved, for example, via sublingual administration (i.e., administration by mouth in such a way that the active ingredient(s) are rapidly absorbed via the
blood vessels under the tongue rather than via the digestive tract). Controlled release formulations (i.e., formulations such as a capsule, tablet or coated tablet that slows and/or delays release of active ingredient(s) following administration) may be administered by, for example, oral, rectal or subcutaneous implantation, or by implantation at a target site. In general, a controlled release formulation comprises a matrix and/or coating that delays disintegration and absorption in the gastrointestinal tract (or implantation site) and thereby provides a delayed action or a sustained action over a longer period. One type of controlled-release formulation is a sustained-release formulation, in which at least one active ingredient is continuously released over a period of time at a constant rate. Preferably, the therapeutic agent is released at such a rate that blood (e.g., plasma) concentrations are maintained within the therapeutic range, but below toxic levels, over a period of time that is at least 4 hours, preferably at least 8 hours, and more preferably at least 12 hours. Such formulations may generally be prepared using well known technology and administered by, for example, oral, rectal or subcutaneous implantation, or by implantation at the desired target site. Carriers for use within such formulations are biocompatible, and may also be biodegradable; preferably the formulation provides a relatively constant level of modulator release. The amount of active ingredient contained within a sustained release formulation depends upon, for example, the site of implantation, the rate and expected duration of release and the nature of the condition to be treated or prevented.
Controlled release may be achieved by combining the active ingredient(s) with a matrix material that itself alters release rate and/or through the use of a controiled-release coating. The release rate can be varied using methods well known in the art, including (a) varying the thickness or composition of coating, (b) altering the amount or manner of addition of plasticizer in a coating, (c) including additional ingredients, such as release-modifying agents, (d) altering the composition, particle size or particle shape of the matrix, and (e) providing one or more passageways through the coating. The amount of modulator contained within a sustained release formulation depends upon, for example, the method of administration (e.g., the site of implantation), the rate and expected duration of release and the nature of the condition to be treated or prevented.
The matrix material, which itself may or may not serve a controlled-release function, is generally any material that supports the active ingredient(s). For example, a time delay material such as glyceryl monosterate or glyceryl distearate may be employed. Active ingredient(s) may be combined with matrix material prior to formation of the dosage form (e.g., a tablet). Alternatively, or in addition, active ingredient(s) may be coated on the surface of a particle, granule, sphere, microsphere, bead or pellet that comprises the matrix material. Such coating may be achieved by conventional means, such as by dissolving the active ingredient(s) in water or other suitable solvent and spraying. Optionally, additional ingredients are added prior to coating (e.g., to assist binding of the active ingredient(s) to the matrix material or to color the solution). The matrix may then be
coated with a barrier agent prior to application of controlled-release coating. Multiple coated matrix units may, if desired, be encapsulated to generate the final dosage form.
In certain embodiments, a controlled release is achieved through the use of a controlled release coating (i.e., a coating that permits release of active ingredient(s) at a controlled rate in aqueous medium). The controlled release coating should be a strong, continuous film that is smooth, capable of supporting pigments and other additives, non-toxic, inert and tack-free. Coatings that regulate release of the modulator include pH-independent coatings, pH-dependent coatings
(which may be used to release modulator in the stomach) and enteric coatings (which allow the formulation to pass intact through the stomach and into the small intestine, where the coating dissolves and the contents are absorbed by the body). It will be apparent that multiple coatings may be employed (e.g., to allow release of a portion of the dose in the stomach and a portion further along the gastrointestinal tract). For example, a portion of active ingredient(s) may be coated over an enteric coating, and thereby released in the stomach, while the remainder of active ingredient(s) in the matrix core is protected by the enteric coating and released further down the GI tract. pH dependent coatings include, for example, shellac, cellulose acetate phthalate, polyvinyl acetate phthalate, hydroxypropylmethylcellulose phthalate. methacrylic acid ester copolymers and zein.
In certain embodiments, the coating is a hydrophobic material, preferably used in an amount effective to slow the hydration of the gelling agent following administration. Suitable hydrophobic materials include alkyi celluloses (e.g., ethylcellulose or carboxymethylcellulose), cellulose ethers, cellulose esters, acrylic polymers (e.g., poly( acrylic acid), poly(methacrylic acid), acrylic acid and methacrylic acid copolymers, methyl methacrylate copolymers, ethoxy ethyl meth aery 1 ate s, cyanoethyl methacrylate. methacrylic acid alkamide copolymer, poly(methyl methacrylate), polyacrylamide, ammonio methacrylate copolymers, aminoaikyl methacrylate copolymer, poly(methacrylic acid anhydride) and glycidyl methacrylate copolymers) and mixtures of the foregoing. Representative aqueous dispersions of ethylceliulose include, for example,
AQUACOAT® (FMC Corp., Philadelphia, PA) and SURELEASE® (Colorcon, Inc., West Point,
PA), both of which can be applied to the substrate according to the manufacturer's instructions.
Representative acrylic polymers include, for example, the various EUDRAGIT® (Rohm America,
Piscataway, NJ) polymers, which may be used singly or in combination depending on the desired release profile, according to the manufacturer's instructions.
The physical properties of coatings that comprise an aqueous dispersion of a hydrophobic material may be improved by the addition or one or more plasticizers. Suitable plasticizers for alkyi celluloses include, for example, dibutyl sebacate, diethyl phthalate, triethyl citrate, tributyl citrate and triacetin. Suitable piasticizers for acrylic polymers include, for example, citric acid esters such as triethyl citrate and tributyl citrate, dibutyl phthalate, polyethylene glycols, propylene glycol, diethyl phthalate, castor oil and triacetin.
Controiled-release coatings are generally applied using conventional techniques, such as by spraying in the form of an aqueous dispersion, if desired, the coating may comprise pores or channels or to facilitate release of active ingredient. Pores and channels may be generated by well known methods, including the addition of organic or inorganic material that is dissolved, extracted or leached from the coating in the environment of use. Certain such pore-forming materials include hydrophilic polymers, such as hydroxyalkylcelluloses (e.g., hydroxypropylmethylcellulose), cellulose ethers, synthetic water-soluble polymers (e.g., polyvinylpyrrolidone, cross-linked polyvinylpyrrolidone and polyethylene oxide), water-soluble polydextrose, saccharides and polysaccharides and alkali metal salts. Alternatively, or in addition, a controlled release coating may include one or more orifices, which may be formed my methods such as those described in US Patent Nos. 3,845,770; 4,034,758; 4,077,407; 4,088,864; 4,783,337 and 5,071,607. Controiled- release may also be achieved through the use of transdermal patches, using conventional technology (see, e.g., US Patent No. 4,668,232).
Further examples of controlled release formulations, and components thereof, may be found, for example, in US Patent Nos. 4,572,833; 4,587,117; 4,606,909; 4,610,870; 4,684,516; 4,777,049;
4,994,276; 4,996,058; 5,128,143; 5,202,128; 5,376,384; 5,384,133; 5,445,829; 5,510, 1 19;
5,618,560; 5,643,604; 5,891,474; 5,958,456; 6,039,980: 6,143,353; 6,126,969; 6,156,342;
6,197,347; 6.387,394; 6,399,096; 6,437,000; 6,447,796; 6,475,493; 6,491 ,950; 6,524,615;
6,838,094; 6,905,709; 6,923,984; 6,923,988; and 6,911,217; each of which is hereby incorporated by reference for its teaching of the preparation of controlled release dosage forms.
In addition to or together with the above modes of administration, an aminopiperidine or related compound (e.g., formulated as a pharmaceutical composition) may be conveniently added to food or drinking water, for administration to humans or non-human animals including companion animals, such as dogs and cats, and livestock. Animal feed and drinking water compositions may be formulated so that the animal takes in an appropriate quantity of the composition along with its diet. It may also be convenient to present the composition as a premix for addition to feed or drinking water.
Aminopiperidines and related compounds are generally present within a pharmaceutical composition at levels providing a therapeutically effective amount upon administration, as described above. Dosage forms providing dosage levels ranging from about 0.1 mg to about 140 mg per kilogram of body weight per day are preferred (about 0.5 mg to about 7 g per human patient per day), with dosages ranging from 0.1 mg to 50 mg, 30 mg or 10 mg particularly preferred. The amount of active ingredient that may be combined with the carrier to produce a single dosage form will vary depending upon the patient to be treated and the particular mode of administration. Dosage unit forms generally contain from about 1 mg to about 500 mg of an active ingredient. It will be understood, however, that the optimal dose for any particular patient will depend upon a variety of factors, including the activity of the specific compound employed; the age, body weight,
general health, sex and diet of the patient; the time arid route of administration; the rate of excretion; any simultaneous treatment, such as a drug combination; and the type and severity of the particular disease undergoing treatment. Dosage units generally contain from about 10 μg to about 500 mg of each active ingredient. Optimal dosages may be established using routine testing and procedures that are well known in the art.
Pharmaceutical compositions provided herein may also contain additional active agents, which can be chosen from a wide variety of molecules and can function in different ways to enhance the therapeutic effects of a MCH receptor modulator, or to provide a separate therapeutic effect that does not substantially interfere with the activity of the MCH receptor modulator. Such optional active agents, when present, are typically employed in the compositions described herein at a level ranging from about 0.01% to about 50% by weight of the composition, preferably 0.1% to 25%, 0.2% to 15, 0.5% to 10% or 0.5% to 5% by weight of the composition. For example, compositions intended for the treatment of obesity and/or eating disorders, such as bulimia nervosa, may further comprise leptin, a leptin receptor agonist, a melanocortin receptor 4 (MC4) agonist, sibutramine, dexfenfluramine, a growth hormone secretagogue, a beta-3 agonist, a 5HT-2 agonist, an orexin antagonist, a neuropeptide Yi or Y5 antagonist, a galanin antagonist, a CCK agonist, a GLP-I agonist, a cannabinoid receptor antagonist {e.g., a CB 3 antagonist) and/or a corticotropin-releasing hormone agonist. Other active ingredients that may be included within the compositions provided herein include antidepressants, inhibitors of dipeptidyl peptidase JV (DPP IV) and/or diuretics. In certain embodiments, an additional active agent is a CBl antagonist. Representative CBl antagonists include, for example, certain pyrimidines {e.g., PCT International Application Publication No. WO 04/029,204), pyrazines {e.g., PCT International Application Publication Nos. WO 01 /1 1 1 ,038; WO 04/1 1 1,034 and WO 04/11 1 ,033). azetidine derivatives {e.g., US Patent Nos. 6,518,264; 6,479,479 and 6,355,631 ; and PCT International Application Publication No. WO 03/053431). pyrazole derivatives {e.g., US Patent Nos. 6,509,367 and 6,476,060; and PCT International Application Publication Nos. WO 03/020217 and WO 01/029007), pyrazolecarboxylic acid and pyrazole carboxamide derivatives {e.g., US patent Nos. 6,645,985; 6,432,984; 6,344.474; 6,028,084; 5,925,768; 5,624,941 and 5,462,960; published US applications US 2004/0039024; US 2003/0199536 and US 2003/0003145; and PCT International Application Publication Nos. WO 03/078413; WO 03/027076; WO 03/026648 and WO 03/026647); aroyl substituted benzofurans {e.g., LY-320135, US Patent No. 5,747,524); substituted imidazoles {e.g., published US application US 2003/0114495 and PCT International Application Publication Nos. WO 03/063781 and WO 03/040107); substituted furo[2,3-b]pyridine derivatives {e.g., PCT International Application Publication No. WO 04/012671); substituted aryl amides {e.g., PCT International Application Publication Nos. WO 03/087037 and WO 03/077847); substituted bicyclic or spirocyclic amides {e.g., PCT International Application Publication Nos. WO 03/086288 and WO 03/082190); and substituted 2,3-diphenyl pyridines {e.g , PCT International Application Publication No. WO
03/08219]). Other CBl antagonists are cannabidiol and its derivatives. Preferred CB l antagonists include, for example, aryl substituted pyrazole carboxamides such as SR-141716A (N-ρiperidin-1- yl)-5-(4-chIoropheny1)-l-(2,4-dichloroρhenyl)-4-methyi- 1 -H-pyrazole-3-carboxamide) as well analogues thereof such as AM251 (N-piperidin-l-yl)-5-(4-iodophenyl)-l-(2,4-dichlorophenyl)-4- methyl- 1 -H-pyrazole-3-carboxamide) and AM281 (N-(morphoIin-4-yl)-] -(2,4-dichioiOphenyl)-5-(4- iodophenyl)-4-methyl-l -H-pyrazole-3-carboxamide); various azetidine compounds (e.g., US Patent Nos. 6,518,264; 6,479,479 and 6,355,631) and the imidazoles l-(4-chlorophenyl)-2-(2- chloroρhenyl)-N-[(l S,2S)-2-hydroxycyclohexyl]-lH-imidazole-4-carboxamide and 2-(2- chlorophenyl)-l-(4-chloropheny!)-N'-[4-(trifluoromethyl)phenyl]-lH-iniidazole-4-cai*bohydrazide. Pharmaceutical compositions may be packaged for treating or preventing a disease or disorder that is associated with MCΗ receptor activation (e.g., treatment of metabolic disorders such as diabetes, heart disease, stroke, metabolic syndrome, obesity and eating disorders such as bulimia, skin disorders such as vitiligo, or sexual disorders such as anorgasmic or psychogenic impotence), or for promoting weight loss. Packaged pharmaceutical preparations comprise a container holding a therapeutically effective amount of MCΗ receptor modulator as described herein and instructions (e.g., labeling) indicating that the contained composition is to be used for promoting weight loss or for treating or preventing a disease or disorder that is associated with MCΗ receptor activation in the patient. Prescribing information may be provided separately to a patient or health care provider, or may be provided as a label or package insert. Prescribing information may include, for example, efficacy, dosage and administration, contraindication and adverse reaction information pertaining to the pharmaceutical formulation. Certain packaged pharmaceutical preparations further include a second therapeutic agent, as discussed above.
METHODS OF USE
Within certain aspects, the present invention provides methods for treating, preventing, or inhibiting the development or progression of a disease or disorder responsive to MCH receptor modulation. In other words, therapeutic methods provided herein may be used to treat a patient already afflicted with such a disease or disorder, or may be used to prevent or delay the onset of such a disease or disorder in a patient who is free of detectable disease or disorder that is associated with MCH receptor activation. As noted above, a disease or disorder is "associated with MCH receptor activation"' if it is characterized by inappropriate stimulation of MCH receptor, regardless of the amount of MCH present locally, and/or is responsive to modulation of MCH receptor activity. Such conditions include, for example, metabolic disorders (such as diabetes), metabolic syndrome, heart disease, stroke, eating disorders (such as obesity and bulimia nervosa), disorders of the skin such as vitiligo, and sexual disorders such as anorgasmic or psychogenic impotence. These conditions may be diagnosed and monitored using criteria that have been established in the art. In addition, MCH antagonists provided herein may be used to promote weight loss in patients, and MCH agonists provided herein may be used to promote weight gain in patients. Patients may
include humans, domesticated companion animals (pets, such as dogs and cats) and livestock animals, with dosages and treatment regimes as described above.
Additional conditions that are associated with MCH receptor activation include: Cognitive impairment and memory disorders, such as Alzheimer's disease, Parkinson's disease. mild cognitive impairment (MCI), age-related cognitive decline (ARCD), stroke, traumatic brain injury, AIDS associated dementia, and dementia associated with depression, anxiety and psychosis (including schizophrenia and hallucinatory disorders);
Anxiety, depression and other mood disorders, including general anxiety disorder (GAD), agoraphobia, panic disorder with and without agoraphobia, social phobia, specific phobia, post traumatic stress disorder, obsessive compulsive disorder (OCD), dysthymia, adjustment disorders with disturbance of mood and anxiety, separation anxiety disorder, anticipatory anxiety acute stress disorder, adjustment disorders and cyclothymia; Reward system disorders such as addiction (e.g., opioid, nicotine or alcohol); Pain such as migraine, peripheral inflammatory pain, neuropathic pain and sympathetic nervous system associated pain; and
Peripheral indications such as respiratory disorders (e.g., asthma), urinary disorders (e.g., urinary incontinence), gastrointestinal disorders, reproductive function disorders and cardiovascular disorders (e.g., arteriosclerosis and hypertension).
Frequency of dosage may vary depending on the compound used and the particular disease to be treated or prevented. In general, for treatment of most disorders, a dosage regimen of 4 times daily or less is preferred. For the treatment of eating disorders and obesity, a dosage regimen of 1 or
2 times daily is particularly preferred. For the treatment of impotence a single dose that rapidly reaches effective concentrations is desirable. It will be understood, however, that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the patient's age, body weight, general health, sex and diet, the time and route of administration, the rate of excretion, any coadministered drugs and the severity of the particular disease. In certain embodiments, administration at meal times is preferred. In general, the use of the minimum dosage that is sufficient to provide effective therapy is preferred. Patients may generally be monitored for therapeutic effectiveness using assays suitable for the condition being treated or prevented, which will be familiar to those of ordinary skill in the art.
In other aspects, methods for treating a patient are provided, comprising diagnosing the patient as having a disease or disorder associated with MCH receptor activation, correlating the diagnosis of the disease or disorder with the need for MCH modulator administration, and administering an a effective amount of an aminopiperidine or related compound provided herein. A method for treating a patient comprising administering an effective amount of an aminopiperidine or related compound to a patient having a disease or disorder associated with MCH receptor activation is also provided herein.
Within certain embodiments the disease or disorder associated with MCH receptor activation is obesity, metabolic syndrome, an eating disorder, a sexual disorder, diabetes, heart disease or stroke.
Within certain embodiments provided herein the aminopiperidine or related compound is administered orally, intranasally, intravenously or topically.
Within certain aspects, MCH receptor modulators provided herein may be used within combination therapy for the treatment of conditions associated with MCH receptor modulation. Within combination therapy, a MCH receptor modulator is administered to a patient along with a second therapeutic agent that is not primarily a MCH receptor modulator, but that is appropriate for treatment of the condition(s) of interest. The MCH receptor modulator and second therapeutic agent(s) may be present in the same pharmaceutical composition, or may be administered separately in either order. Suitable second therapeutic agents include those listed above.
Suitable dosages for MCH receptor modulator(s) within such combination therapy are generally as described herein. Dosages and methods of administration of other therapeutic agents can be found, for example, in the manufacturer's instructions in the Physician's Desk Reference. In certain embodiments, the combination administration results in a reduction of the dosage of the second therapeutic agent required to produce a therapeutic effect {i.e., a decrease in the minimum therapeutically effective amount). Thus, preferably, the dosage of second therapeutic agent in a combination or combination treatment method of the invention is less than the maximum dose advised by the manufacturer for administration of the second therapeutic agent without combination administration of a MCH receptor modulator. More preferably this dosage is less than 3Zi, even more preferably less than 14, and highly preferably, less than VA of the maximum dose, while most preferably the dose is less than 10% of the maximum dose advised by the manufacturer for administration of the second therapeutic agent(s) when administered without combination administration of a MCH receptor modulator. It will be apparent that the dosage amount of MCH receptor modulator component of the combination needed to achieve the desired effect may similarly be affected by the dosage amount and potency of the second therapeutic agent component of the combination.
In certain preferred embodiments, the combination administration of a MCH receptor modulator with a second therapeutic agent is accomplished by packaging one or more MCH receptor modulators and one or more second therapeutic agents in the same package, either in separate containers within the package or in the same container as a mixture of one or more MCH receptor modulators and one or more second therapeutic agents. Preferred mixtures are formulated for oral administration {e.g., as pills, capsules, tablets or the like). In certain embodiments, the package comprises a label or package insert indicating that the one or more MCH receptor modulators and one or more second therapeutic agents are to be taken together for the treatment of a condition that is associated with MCH receptor activation, such as obesity.
In certain embodiments, one or more MCH receptor modulators provided herein are used along with one or more CBl antagonists within a combination therapy. Such combinations are of particular use for weight management, to reduce appetite and/or food intake or to prevent or treat obesity (e.g., promote weight loss). Patients may include humans, domesticated companion animals and livestock animals, with dosages and treatment regimes as described above. The MCH receptor modulator(s) may be administered to the patient at the same time as the CBl antagonist(s) (e.g., as a single dosage unit), or may be administered separately (before or after CBl antagonist). Within preferred embodiments, the MCH receptor modulators) and CBl antagonist s) are ultimately simultaneously present at effective concentrations in a body fluid (e.g., blood) of the patient. An effective concentration of MCH receptor modulator or CBl antagonist is a concentration that is sufficient to reduce one or more of food consumption, appetite and/or body mass index in the patient when repeatedly coadministered as described herein.
Within separate aspects, the present invention provides a variety of in vitro uses for the compounds provided herein. For example, such compounds may be used as probes for the detection and localization of MCH receptors, in samples such as tissue sections, as positive controls in assays for receptor activity, as standards and reagents for determining the ability of a candidate agent to bind to MCH receptor, or as radiotracers for positron emission tomography (PET) imaging or for single photon emission computerized tomography (SPECT). Such assays can be used to characterize MCH receptors in living subjects. Compounds provided herein are also useful as standards and reagents in determining the ability of a test compound to bind to MCH receptor.
Within methods for determining the presence or absence of MCH receptor in a sample, a sample may be incubated with a compound as provided herein under conditions that permit binding of the compound to MCH receptor. The amount of compound bound to MCH receptor in the sample is then detected. For example, a compound may be labeled using any of a variety of well-known techniques (e.g., radiolabeled with a radionucleide such as tritium, as described herein), and incubated with the sample (which may be. for example, a preparation of cultured cells, a tissue preparation or a fraction thereof). A suitable incubation time may generally be determined by assaying the level of binding that occurs over a period of time. Following incubation, unbound compound is removed, and bound compound detected using any method for the label employed (e.g., autoradiography or scintillation counting for radiolabeled compounds; spectroscopic methods may be used to detect luminescent groups and fluorescent groups). As a control, a matched sample may be simultaneously contacted with radiolabeled compound and a greater amount of unlabeled compound. Unbound labeled and unlabeled compound is then removed in the same fashion, and bound label is detected. A greater amount of detectable label in the test sample than in the control indicates the presence of MCH receptor in the sample. Detection assays, including receptor autoradiography (receptor mapping) of MCH receptors in cultured cells or tissue samples may be
performed as described by Kuhar in sections 8.1.1 to 8.1.9 of Current Protocols In Pharmacology (1998) John Wiley & Sons, New York.
Compounds provided herein may also be used within a variety of well-known cell culture and cell separation methods. For example, compounds may be linked to the interior surface of a tissue culture plate or other cell culture support, for use in immobilizing MCH receptor-expressing cells for screens, assays and growth in culture. Compounds may also be used to facilitate ceil identification and sorting in vitro, permitting the selection of cells expressing a MCH receptor.
Preferably, the compound(s) for use in such methods are labeled as described herein. Within one preferred embodiment, a compound linked to a fluorescent marker, such as fluorescein, is contacted with the cells, which are then analyzed by fluorescence activated cell sorting (FACS).
Within other aspects, methods are provided for modulating binding of MCH to an MCH receptor in vitro or in vivo, comprising contacting a MCH receptor with a sufficient amount of a modulator provided herein, under conditions suitable for binding of MCH to the receptor. Preferably, within such methods, MCH binding to receptor is inhibited by the modulator. The MCH receptor may be present in solution, in a cultured or isolated cell preparation or within a patient. Preferably, the MCH receptor is a MCHlR receptor present in the hypothalamus. In general, the amount of compound contacted with the receptor should be sufficient to modulate MCH binding to MCH receptor in vitro within, for example, a binding assay as described in Example 6 and/or Example 9. MCH receptor preparations used to determine in vitro binding may be obtained from a variety of sources, such as from HEK 293 cells or Chinese Hamster Ovary (CHO) cells transfected with a MCH receptor expression vector, as described herein.
Also provided herein are methods for modulating the signal-transducing activity of cellular MCH receptors, by contacting MCH receptor, either in vitro or in vivo, with a sufficient amount of a modulator as described above, under conditions suitable for binding of MCH to the receptor. Preferably, within such methods, signal-transducing activity is inhibited by the modulator. The MCH receptor may be present in solution, in a cultured or isolated cell preparation or within a patient. In general, the amount of modulator contacted with the receptor should be sufficient to modulate MCH receptor signal transducing activity in vitro within, for example, a calcium mobilization assay as described in Example 30 and/or an agonist-stimulated GTP gamma35S binding assay as described in Example 8. An effect on signal-transducing activity may be assessed as an alteration in the electrophyϋiology of the cells, using standard techniques, such as intracellular patch clamp recording or patch clamp recording. If the receptor is present in an animal, an alteration in the electrophysiology of the cell may be detected as a change in the animal's feeding behavior.
PREPARATION OF AMINOPIPERIDINES AND RELATED COMPOUNDS Aminopiperidines and related analogues provided herein may generally be prepared using standard synthetic methods. Starting materials are generally readily available from commercial
sources, such as Sigma-Aldrich Corp. (St. Louis, MO). For example, a synthetic route similar to that shown in any one of the following Schemes may be used. It will be apparent that the finai product and any intermediate(s) shown in the following schemes may be extracted, dried, filtered and/or concentrated, and may be further purified (e.g., by chromatography). Each variable (e.g , "R") in the following Schemes, refers to any group consistent with the description of the compounds provided herein. An individual skilled in the art may modify one or several of the synthetic steps described herein without diverting significantly from the overal! synthetic scheme. Further experimental details for synthesis of representative compounds via these schemes are provided in Examples 1 -3, herein. In the following Schemes and elsewhere herein, the following abbreviations are used:
AcOH acetic acid
BOP benzotriazol- 1 -yloxy-tris(dimethylamino)phosphonium hexafluorophosphate
DCE 1,2-dichloroethane
DCM dichloromethane DMA N,N-dimethylacelamide
DMAP 4-dimethyiaminopyridine
DMC 2-chloro-l,3-dimethylimidazolinium chloride
DMSO dimethyl sulfoxide
DMF dimethylformamide DPPP l,3-bis(diphenyl-phosphino)propane
DPPF 3 , 1 '-bis(diphenyiphosρhino)ferrocene
EDCI 3 ' -(dimethylaminopropyi)carbodiimide
Eq. equivalent(s)
EtOAc ethyl acetate EtOH ethanol h hour(s)
HBTU 0-( 1 H-benzotriazol-yli-ΝjΝ^'.Ν'-tetramethyluronium iPrOH isopropanol
MeOH methanol Me-THF 2-methyl tetrahydrofuran min minute(s)
OAc acetate
OiPr isopropoxy
Pd2(dba)3 tris[dibenzylidineacetonejdi-ρalladium PPh3 triphenylphosphine
PPTS pyridinium-jcαrø-toluene sulfonic acid
PTLC preparative thin layer chromatography
Vt room temperature
SCX strong cation exchange
TBAF tetra-n-butyiammonium fluoride
TBS tert-buty] dimethylsilyl
TEA triethylamine
THF tetrahydrofuran
TLC thin layer chromatography
TR retention time
SCHEME 1
4M HCI/dioxaπe EtOAc



SCHEME 3
HCI/ciioxane EtOAc

SCHEME 4
4M HC!/d(oxane EtOAc

RACOOH
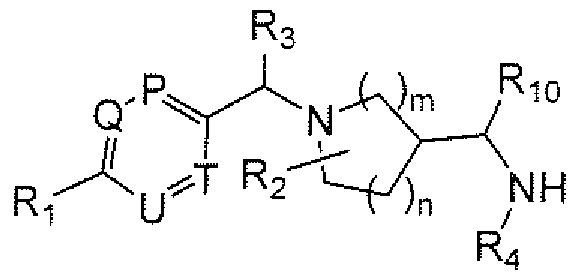


SCHEME 5

SCHEME 6

SCHEME 7

Pd(OAc)2, dppf, CO MeOH, TEA, DMF


In certain situations, compounds provided herein may contain one or more asymmetric carbon atoms, so that the compounds can exist in different stereoisomeric forms. These compounds
can be, for example, racemates or optically active forms. As noted above, ail stereoisomers are encompassed by the present invention. Nonetheless, it may be desirable to obtain single enantiomers (Le., optically active forms). Standard methods for preparing single enantiomers include asymmetric synthesis and resolution of the racemates. Resolution of the racemates can be accomplished, for example, by conventional methods such as crystallization in the presence of a resolving agent, or chromatography using, for example, a chiral HPLC column. As noted above, for compounds having an alpha-methyl benzyl group (R3 is methyl) the R enantiomer is generally preferred.
Compounds may be labeled by carrying out their synthesis using precursors comprising at least one atom that is an isotope. Each isotope is preferably carbon (e.g., 14C), hydrogen (e.g., 3H or
2H), fluorine (e.g., I SF), sulfur (e.g., 35S) or iodine (e.g., 125T). Tritium labeled compounds may also be prepared catalytically via platinum-catalyzed exchange in tritiated acetic acid, acid-catalyzed exchange in tritiated trifluoroacetic acid, or exchange with tritium gas under heterogeneous catalysis using the compound as substrate. In addition, certain precursors may be subjected to tritium- halogen exchange with tritium gas, tritium gas reduction of unsaturated bonds, or reduction using sodium borotritide, as appropriate. Preparation of radiolabeled compounds may be conveniently performed by a radioisotope supplier specializing in custom synthesis of radiolabeled probe compounds.
The following Examples are offered by way of illustration and not by way of limitation. Unless otherwise specified, all reagents and solvent are of standard commercial grade and are used without further purification. Starting materials are available from commercial suppliers, such as Sigma-Aldrich (St. Louis, MO), or are synthesized using procedures that are known in the art.
EXAMPLES Mass spectroscopy in the following Examples is Electrospray MS, obtained in positive ion mode using a Micromass Time-of-Flight LCT (Waters Corp.; Milford, MA) (Methods 1 and 2) or a
Waters ZMD II Mass Spectrometer (Method 3), equipped with a Waters 600 pump (Waters Corp.;
Milford, MA), Waters 996 photodiode array detector (Waters Corp.; Milford, MA), and a Gilson
215 autosampler (Gilson, Inc.; Middleton, WI). MassLynx™ (Waters Corp.; Milford, MA) version 4.0 software with OpenLynx Global Server™, OpenLynx™ and AutoLynx™ processing is used for data collection and analysis.
HPLC and MS conditions are as follows. Unless otherwise specified, mass spectroscopy data in the following Examples is obtained via HPLC MS Method 1. Where a different method is used, the method is indicated.
Method 1
MS conditions are as follows: capillary voltage = 3.5 kV; cone voltage = 30 V, desolvation and source temperature = 35O0C and 12O0C, respectiveiy; mass range = 181 -750 with a scan time of 0.22 seconds and an InterScan delay of 0.05 seconds.
Sample volume of 1 microliter is injected onto a 50x4.6mm Chromolith SpeedROD RP-I Se column (Merck KGaA, Darmstadt, Germany), and eluted using a 2-ρhase linear gradient at a flow rate of 6 ml/min. Sample is detected using total absorbance count over the 220-340nm UV range. The elution conditions are: Mobile Phase A - 95% water, 5% MeOH with 0.05% TFA; Mobile Phase B - 5% water, 95% MeOH with 0.025% TFA. The following gradient is used: 0-0.5 min 10- 100%B, hold at ϊ 00%B to 1.2 min, return to 10%B at 1.2 ] min. Inject to inject cycle is 2.15 min.
Method 2
The conditions and procedure are the same as for Method 1, except that the following gradient is used: 0-0.5 min 5-300%B, hold at 100%B to 1.2 min, return to 5%B at 1.21 min. Inject to inject cycle is 2.15 min. Method 3
MS conditions are as follows: capillary voltage = 3.5 kV; cone voltage ~ 30 V, desolvation and source temperature = 25O0C and 12O0C, respectively; mass range = 100-750 with a scan time of 0.5 seconds and an interscan delay of 0.1 seconds.
Sample volume of 1-10 microliter is injected onto a 30x4.6mm XBridge Cl 8 5 micron column or equivalent, and eluted using a 2-phase linear gradient at a flow rate of 4 ml/min. Sample is detected using total absorbance count over the 220-254 UV range. The elution conditions are: Mobile Phase A - 95% water, 5% MeOH with 0.1% ammonium hydroxide; Mobile Phase B - 5% water, 95% MeOH with 0.025% formic acid. The following gradient is used: 0.01-2.00 min 0- 100% B, hold at 100%B to 3.50 min, return to 0%B at 3.51 min.
EXAMPLE 1. PREPARATION OF REPRESENTATIVE COMPOUNDS
This Example illustrates the preparation of certain representative aminopiperidines and related compounds.
1 . 3-CHLORO-4-FLUORO-N- {8-[4-(2 -METHOXYETHOXY)^5S-DIMETHYLBENZYL]- ΛZABICYCLO[3.2.1 ]OCT-3- YL}BENZAMIDE


Nortropinone (prepared essentially as described by Berdini et a!. (2002) Tetrahedron 58:5669; 1.50 g, 12.0 mmol), and 2,3-dimethyI-4-ethoxy(2-methoxy)benzaIdehyde (2.71 g, 13.0 mmol) are dissolved in dry CH2Cl? (100 mL). Sodium triacetoxyborohydride (3.56 g, 17,0 mmol) is added and the mixture is stirred overnight. The reaction is quenched by the addition of 1 N NaOH solution (50 mL) and the resulting layers are separated. The aqueous iayer is extracted with CH2Cl2 (2 x 50 mL), and the combined organic layers are dried over Na2SC^ and evaporated to yield crude intermediate. The crude intermediate is dissolved in pyridine (20 mL), and water (10 mL) and hydroxylamine hydrochloride (0.92 g, 13.3 mmol) are added. The solution is stirred for 6 h. The volatiles are evaporated and the product is partitioned between 1 N NaOH (50 mL) and EtOAc (50 mL), The layers are separated, and the aqueous layer is extracted with EtOAc (2 x 25 mL). The combined organic layers are washed with brine, dried over Na2SO4 and evaporated. Ether (10 mL) is added and the solution is placed in a refrigerator overnight. The resulting precipitate is collected by vacuum filtration to give the title compound as a white solid. 1H NMR (300 MHz. DMSO-d6) δ 10.24 (s, 1 H), 7.03 (d, 1 H), 6.69 (s, 1 H), 4.02 (t, 2 H), 3.65 (t, 2 H), 3.47 (s, 2 H), 3.31 (s, 3 H), 3.20 (d, 2 H). 2.80 (d, 1 H), 2.35 (d, 1 H), 2.24 (s, 3 H), 2.09 (s, 3 H), 2.00-1.90 (m, 4 H), 1.43 (t, 1 H), 1.29 (t, I H). m/z 333.21 (M+ 1).
Step 2, 8-[4-(2-Methoxy-ethoxy)-2,3-dimethy l-benzyl]-8-aza-bicycIo[3.2.1]oct-3-ylamine

8-[4-(2-Methoxy-ethoxy)-2,3-dimethyl-benzyi]-8-aza-bicycIo[3.2.1]octan-23-one oxime (1.00 g, 3.47 mmol) is dissolved in dry n-propanol (20 mL) and heated to 80 0C, at which point sodium metal (0.80 g. 34.7 mmol) is added over a 2 min period. Once the addition is complete, the solution is heated to 125 0C and stirred for 2 h. The solution is cooled, water (40 mL) is added, the pH is adjusted to 9 by the addition of 6 N HCl, and the solution is transferred to a separatory funnel. The product is extracted into EtOAc (3 x 30 mL). The combined organic layers are washed with brine, dried over Na2SO4 and evaporated to give the title compound as a light yellow syrup. 1H NMR (300 MHz5 DMSO-d6) d 7.94 (bs, 2 H), 6.98 (d, 1 H), 6.70 (d, 1 H), 4.03-4.00 (m, 2 H), 3.67- 3.63 (m, 2 H), 3.42 (bs, 2 H), 3.31 (s. 3 H), 3.28-3.09 (m, 3 H), 2.22 (s, 3 H), 2.09 (s, 3 H), 1.99-1.90 (m, 2 H), 1.75-1.48 (m, 6 H). m/z 319.24 (M+l).
Step 3. 3-CHLORO-4-FLUORO-N- {8-[4<2-METHOXYETHOXY)-2,3-DIMETHYLBENZYL]-8- ΛZABICYCL0[3.2. ϊ ]0CT-3-YL}BENZAMIDE

8-[4-(2-Methoxy-ethoxy)-2,3-dimethy l-benzylJ-δ-aza-bicycloP^. ljoct-B-ylamine (0.30 g, 0.94 mmol) and 3-chloro-4-fluorobenzoic acid (0.19 g, 1.1 mmol) are dissolved in dry toluene (10 mL). TEA (0.38 mL, 2.8 mmol) and BOP (0.84 g, 1.9 mmol) are added and the solution is heated to 50 0C for 3 h. The solution is cooled, 1 N NaOH (20 mL) is added, and the solution is extracted with ether (3 x 20 mL). The combined extracts are washed with brine, dried over Na2SO4 and evaporated. The residue is applied to a solid phase extraction column (LC-SCX), contaminants are removed by elution with 10% MeOH in EtOAc, and the product is collected by elution with 10: 1 : 1 EtOAc:MeOH:TEA, and evaporated to give the title compound. 1H NMR (300 MHz, CDCl3) δ 7.83-7.56 (series of multiplets, 1 H), 7.18-6.99 (series of multiplets, 2 H), 6.65 (d, 1 H), 6.06-5.87 (series of multiplets, 1 H), 4.36-4.20 (m, 1 H), 4.09 (dd, 2 H), 3.77 (dd, 2 H), 4.33 (s, 3 H), 3.23 (bs, 3 H), 2.88-2.77 (series of multiplets, 2 H), 2.30 (s, 3 H), 2.20 (s, 3 H), 2.09-2.05 (m, 2 H), 1.90-1.84 (111, 2 H), 1.78-1.74 (m, 2H)5 3.63-1.55 (m, 2 H). m/∑ 475.28 (M+ 1).
2. 3-CHLORO-4-FLUORO-N-[8-(4-METHOXY-2,3-DIMETHYLBENZYL)-8-AZABICYCLO[3.2.1 ]OCT-3- YL]BENZAMIDE

This compound is prepared a described for Compound 1, with readily apparent modification of starting materials.
3. 3-CHLORO-N-(8-{4-[3-(DIMETHYLAMINO)PROPOXY]-2,3-DIMETHYLBENZYL}-8- AZAB IC YCLO[3.2.1] OCT-3 - YL)-4-FLUOROBENZAMIDE


3-ChloiO-4-fluoro-N-[8-(4-methoxy-2,3-dimethy!benzyl)-8-azabicyclo[3.2.1]oct-3- yljbenzamide (1.20 g. 2.8 mmol) is dissolved in EtOAc (I O mL). HCl (1 M in diethyl ether, 2.8 mL, 2.8 mmol) is added, and the resulting precipitate is collected by vacuum filtration. The salt (0.20 g, 0.43 mmol) is suspended in dry CHbCI2. and cooled to -78 0C, and neat boron tribromide (0.10 mL, 1 .0 mmol) is added. The mixture is stirred for 30 min, the cold bath is removed and the solution is stirred for another 2 h. The reaction is quenched by the addition of saturated sodium bicarbonate solution (10 mL). The layers are separated, and the aqueous layer is extracted with CH2Cl2 (2 x 5 mL). The combined extracts are washed with brine, dried over Na2SO4 and evaporated, to give the title compound as a light yellow syrup. 1H NMR (300 MHz, CDCl3) δ 7.83-7.56 (series of multiples, 1 H), 7.18-6.99 (series of multiplets, 2 H), 6.65 (d, I H), 6.06-5.87 (series of multiplets, 1 H), 4.36-4.20 (m, 1 H), 3.42 (s, 2 H), 3.18-3.3.16 (m, 2 H), 2.30 (s, 3 H)5 2.20 (s, 3 H). 2.01-1.98 (m, 2 H). 1 .71-1.66 (m, 2 H), 1.58-1.55 (m, 2 H), 1.45-1.32 (m. 2 H), OH, and NH not observed, m/z 417.21 (M+] ).
Step 2. 3-Chloro-N-(8-{4-[3-chloro-propoxy]-2,3-dimethylbenzyl}-8-azabicyclo[3.2.1]oct-3-yl)-4- fluorobenzamide


3-Chloro-N-(8-{4-[3-chloro-propoxy]-2.3-dimethylbenzyI}-S-azabicyclo[3.2. I]oct-3-yl)-4- fluorobenzamide (0.062 g. 0.13 mmol) is dissolved in DMA (0.4 mL). Potassium carbonate (0.054 g, 0.39 mmol) is added, followed by dimethylamine-HCl (0.021 g, 0.26 mmol). The mixture is heated to 70 0C for 18 h, the solvent is evaporated and the residue is purified by PTLC (10:1 :0.1, CH2Cl2: MeOH: NH4OH) to give the title compound as a colorless syrup. 1H NMR (300 MHz, CDCl3) δ 7.82-7.56 (series of multiplets, 1 H), 7.19-6.99 (series of muitiplets, 2 H), 6.64 (d, 1 H), 5.86 (dd, 1 H). 4.39-4.24 (m, 1 H), 3.98 (t, 2 H), 3.45 (s, 2 H), 3.24 (bs, 2 H), 2.86 (s, 2 H), 2.48 (t, 3 H), 2.31 (s, 3 H), 2.26 (s, 6 H), 2.18 (s, 3 H), 2.09-1.52 (series of multiplets, 8 H). m/z 502.21 (M-H).
4. 3-CHLORO-N-(8-{4-[3-(METΠYLAMINO)PROPOXY]-2,3-DIMETHYLBENZYL}-8-
AZΛBICYCLO[3.2. IJOCT-J-YLH-FLUOROBBNZAMIDE

Reaction of 3-chloro-N-(8-{4-[3-chloro-propoxy]-2,3-dimethylbenzy!}-8-azabicyclo- [3.2.1]oct-3-yl)-4-ftuorobenzamide (0.035 g, 0.07 mmol) with methylamine (2 M in THF, 0.07 mL, 0.14 mmol) and potassium carbonate (0.012 g, 0.09 mmol) in DMA (0.30 mL), as described for Compound 3, gives the title compound as a colorless syrup. 1H NMR (300 MHz, CDCl3) δ 7.75-7.49 (series of multiplets, 1 H), 7.13-6.93 (series of multiplets, 2 H), 6.61 (dd, 1 H), 5.80 (dd, 1 H), 4.63- 4.30 (m, 1 H), 3.96 (t, 2 H). 3.77 (s. 2 H), 3.42 (s. 2 H), 3.23 (bs, 2 H). 2.82-2.76 (m, 4 H), 2.43-1.53 (series of multiplets, 15 H), NH not observed, m/z 488.23 (M+ 1).
5. 3 -CHLORO-4-FLUORO-N -[8-(4- { 3 - [(2-HYDROXYETHYL)AMINO]PROPOXY } 2,3 - DIMETHYLBENZYL)-S-AZABICYCLO[S .2. l ]OCT-3 -YL]BENZAJVI1DE

6. N-(8-{4-[(R)-2-(TERT-BUTYL-DJMETHYL-SILΛNYLOXY)-PROPOXY]-2,3-DIMETHYL-BENZYL}-8- AZA-BICYCLOp .2.1 ]OCT-3 -YL) -3-CHLORO^-FLUORO-BENZAMIDE

3-Chloro-4-fluoro-N-[8-(4-hydroxy-2,3-dimethylbenzyi)-8-azabicycio[3.2.13oct-3- yljbenzamide (0.104 g, 0.24 mmol) is dissolved in diy DMF (8.0 mL). (R)-propyIeneglycol-2-ter/- butyldimethylsilyloxy-1-tosylate (0.15 g, 0.42 mmol) is added, followed by cesium carbonate (0.20 g, 0.60 mmol), and the resulting mixture is heated to 60 0C for 36 h. The solution is cooled, water (15 mL) is added and the solution is extracted with EtOAc (3 x 20 mL). The combined organic extracts are washed with water (10 mL) and brine (15 mL), dried over Na2SO4 and evaporated. The residue is purified by SPE-SCX, yielding the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.81-7.56 (series of multiplets, 1 H), 7.21-7.00 (series of multiplets, 2 H), 6.64 (d, 1 H), 5.92 (dd. 1 H), 4.39-4.15 (series of multiplets, 2 H), 3.95-3.77 (m, 2 H), 3.56 (s, 2 H), 3.33 (bs, 2 H), 2.86 (s, 2 H), 2.32 (s, 3 H), 2.20 (s 3 H), 2.13-1.1 1 (series of multiplets, 10 H), 0.90 (s, 9 H), 0.10 (s. 3 H), 0.08 (s, 3 H). m/z 589.82 (M+I ).
7. N-(8-{4-[(S)-2-(r£Λr-BUTYL-DIMETHYL-SILANYLOXY)-PROPOXY]-2,3-DIMETHYL-BENZYL}-8- AZA-BIC YCLO[3.2.1 ]OCT-3- YL)-3-CHLORO-4-FLUORO-BENZAMIDE

Treatment of 3-chloro-4-ffuoro-N-[8-(4-hydroxy-2,3-dimethylbenzyl)-8-azabicyclo[3.2.1] oct-3-y]]benzamide (0.21 g, 0.5 mmol) with (S)-propyIenegIycol-2-tert-butyldimethy!silyloxy-l-
tosylate (0.30 g, 0.86 mmoi), as described for Compound 6, gives the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.81-7.56 (series of multiplets, 1 H), 7.21-7.00 (series of multiplets, 2 H), 6.64 (d, 1 H), 5.92 (dd, 1 H), 4.39-4.15 (series of multiplets, 2 H), 3.95-3.77 (m, 2 H), 3.56 (s, 2 H), 3.33 (bs, 2 H), 2.86 (s, 2 H), 2.32 (s, 3 H), 2.20 (s 3 H), 2.13-1.1 1 (series of multiplets, 10 H), 0.90 (s, 9 H), 0.10 (s, 3 H), 0.08 (s, 3 H). m/z 589.81 (M+l).
8. 3-CHLORO-4-FLUORO-N- {8-[4-((R)-2-HYDROXY-PROPOXY)-2,3-DIMETHYL-BENZYL]-8~AZA- BICYCLO[3.2.1]OCT-3 -YL}-BENZAMIDE

N-(8-{4-[(R)-2-(fer/-butyl-dimethyl-silanyloxy)-propoxy]-2,3-dimethyl-benzyl}-8-aza- bicyclo[3.2.1]oct-3-yl)-3-chloro-4-fluoro-benzamide (0.06 g, 0.15 mmoi) is dissolved in dry THF (10 ml), cooled to 0 0C, and TBAF (IM in THF, 0.15 mL, 0.15 mmoi) is added. The solution is allowed to warm to rt, stirred for a further 3 h, and the volatiles are evaporated. The residue is purified by PTLC (10% MeOH in CH2CI2) to give the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.81-7.56 (series of multiplets, 1 H), 7.19-7.00 (series of multiplets, 2 H), 6.64 (d, 1 H), 5.85 (dd, 1 H), 4.39-4.15 (series of multiplets, 2 H), 3.92 (dd, 1 H), 3.79 (dd, 1 H), 3.47 (s, 2 H), 3.25 (bs, 2 H), 2.86 (s, 2 H), 2.32 (s, 3 H), 2.20 (s 3 H), 2.13-2.06 (m, 2 H), 1.92-1.86 (m, 2 H), 1 .80-1 .73 (m, 2 H), 1.64-1.57 (m, 2 H), 1 .29 (d, 3 H). m/z 475.45 (M+l).
9. 3-CHLORO-4-FLUORO-N- {8-[4-((S)-2-HYDROXY-PROPOXY)-2,3-DIMETHYL-BENZYL]-8-AZA- BICYCLOp .2.1 ]OCT-3 -YL } -BENZAMIDE

Treatment of N-(8-{4-[(S)-2-(rert-butyl-dimethyl-siIanyioxy)-pi'opoxy]-2,3-dimethyl- benzyl}-8-aza-bicyclo[3.2.1]oct-3-yl)-3-chloro-4-fluoiO-benzamide (0.129 g, 0.20 mmoi) as described in Example IH with TBAF (I M in THF, 0.30 mL, 0.30 mmoi) gives the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.81 -7.56 (series of multiplets, 1 H), 7.19-7.00 (series of multiplets, 2 H), 6.64 (d, 1 H), 5.85 (dd, 1 H), 4.39-4.15 (series of multiplets, 2 H), 3.92 (dd, 1 H), 3.79 (dd, 1 H)5 3.48 (s, 2 H), 3.25 (bs, 2 H), 2.86 (s, 2 H), 2.32 (s, 3 H), 2.20 (s 3 H), 2.13-2.06 (m, 2 H), 1.92-1.86 (m, 2 H), 1.80-1.73 (in, 2 H), 1.64-1.57 (m, 2 H), 1.29 (d, 3 H). m/z 475.44 (M+l).
10. N-(I -( 1 -[4-(ALL YLOXY)-2,3-DIMETHYLPHENYL]ETHYL} PIPERIDIN-4-YL)-2- CHLORON ICOTIN AMJDE
Step 1 . l -(4-methoxy-2,3-dimethyiphenyl)ethanone

Step 2. 1 -(4-hydroxy-2, 3-dimethylphenyl)ethanone
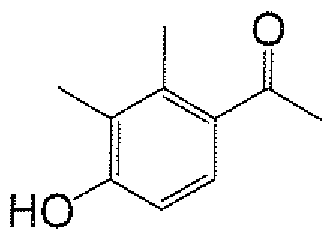
To a solution of l-(4-methoxy-2, 3-dimethyiphenyl)ethanone (37 g, 0.208 moi) in anhydrous CH2CI2 (400 mL) under N2 at -78 0C is added BBr3 (49.2 mL, 0.52 mol) dropwise via an addition funnel over a period of 45 min while maintaining the internal temperature below -70 0C. The reaction mixture is gradually warmed to rt and stirred overnight. The reaction mixture is poured carefully into saturated NaHCO3 solution (3500 mL) containing ice over 30 min with vigorous stirring, and warmed to rt gradually. The pH of the aqueous layer is about 6-7. The light pink solid is collected via filtration and washed with water. The solid is redissolved in EtOAc (500 mL), washed with water and brine, and dried over Na2SO4. Removal of the solvent under reduced pressure affords the title compound as a light pink solid. The organic layer from the filtration of the solid is separated, and the aqueous phase is extracted with CH2Cl2 (2 x 100 mL). Organic layers are combined, washed with water (2 x 250 mL), brine (250 mL), dried over Na2SO4, and concentrated under reduced pressure. The residue is triturated with CH2CWEt2O (1 : 1, 50 mL) to afford additional title compound as a light pink solid. 1H-NMR (400 MHz, DMSO-<i6) δ: 9.96 (s, IH), 7.49 (d, IH), 6.70 (d, I H), 2.43 (s, 3H), 2.29 (s, 3H), 2.05 (s, 3H). LC-MS m/z (M+H): 164.
Step 3. l-[4-(allyloxy)-2, 3-dimethyIphenyl]ethanone

To a solution of l-(4-hydroxy-2,3-dimethylρhenyl)ethanone (26.26 g, 0.161 moi) in anhydrous acetonitrile (300 niL) under N2 at rt is added powdered KOH (9.92 g, 0.177 mol). After stirring at rt for 10 min, allyl iodide (19.1 mL, 0.209 mol) is added, and the reaction mixture is stirred at rt overnight. Acetonitrile is removed under reduced pressure. The residue is diluted with
EtOAc, washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure.
The residue is purified by silica gel chromatography (hexane/EtOAc 95:5) to afford the title compound as a yeilow oil. 1H-NMR (300 MHz, CDCl3) & 7.51 (d, IH), 6.70 (d, IH), 6.07 (m, IH), 5.44 (m, IH), 5.30 (m, IH), 4.57-4.59 (m, 2H), 2.54 (s, 3H), 2.43 (s, 3H), 2.21 (s, 3H). LC-MS m/z
(M+H): 205.
Step 4. !-{ l-[4-al]yloxy)-2,3-dimethylphenyi]ethyl}piperidin-4-amine
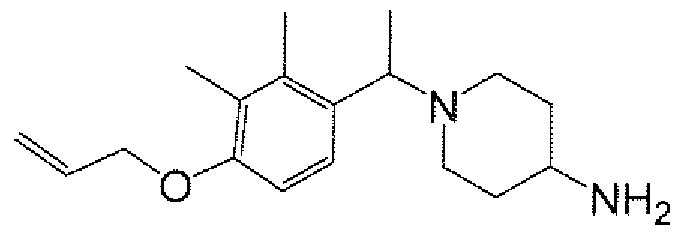

].| i.[4^a]]yjOXj,).2,3-dimetIiylplienyl]ethyi}pipeπdin-4-amine (498 mg, 1.72 rnmol) is dissolved in 5 mL DCM and treated with 2-chloronicotinoyl chloride (365 mg. 2.07 mmol, 1.2 eq.) in 2 mL of DCM. The soiution is stirred at rt for 72 h. The reaction mixture is diluted with 10 mL DCM and washed with half-saturated NaHCU3. The aqueous phase is extracted with DCM and the combined organic extracts are concentrated in vacuo. The residue is purified by silica gel chromatography (2.5-6% MeOH (2M NH3)/DCM to provide the title compound as a white foam. 1H NMR: δ 8.44 (1H), 8.07 (1H), 7.33 (IH)5 7.18 (I H), 6.69 (IH), 6.36 1 (H), 6.08 (IH), 5.45 (IH), 5.27 (IH), 4.51 (2H), 4.00 (IH), 3.65 (IH), 2.93 (IH), 2.74 (IH), 2.28 (3H), 2.21 (3H), 2.19 (IH), 2.03 (1 H), 1.94 (1 H). 1.54 (2H), 1.29 (3H); LCMS: TR = 2.01 min, m/z 428.09 (M+l).
1 1 . N-(l -{ l -[4-(ALLYLOXY)-2,3-DIMETHYLPHENYL]ETHYL}PIPERiDIN-4-YL)-2- (1 SOPROP YLAMINO)N ICOTIN AMIDE

12. 2,5-DICHLORO-7V-{ l -[l-(4-METHOXY-2,3-DIMETHYLPHENYL)El HYL]PΪPERIDIN-4- YL}NICOTIN AMIDE
Step 1 . 1 -[ 1 -(4-methoxy-2,3-dimethylphenyI)ethyl]piperidin-4-amine

A mixture of l-(4-methoxy-2, 3-dimethylphenyl)ethanone (3 g, 0.01685 mol, 1 eq.) and 4- Boc-aminopiperidine (3.3 g, 0.0168 mol, 1 eq.) in Ti(OiPr)4 (10.1 mL, 0.0336 mol, 2 eq.) is heated under N2 at 70 0C for 3 h. The reaction mixture is cooled to 0 0C, and anhydrous EtOH (50 mL) is
added, followed by NaBH4 (0.954 g, 0.0252 mol, 1.5 eq.) portionwise. The mixture is stirred at 0 0C for 1 h and then at rt overnight. The reaction is quenched by addition of aq. NaOH (IN, 75 mL), and stirred at rt for 0.5 h. Insoluble materials are removed by filtration through Celite®, and the filter cake is washed with EtOAc. The filtrate and wash are combined and concentrated under reduced pressure. The residue is partitioned between water and EtOAc. The organic layer is washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure to afford a yellow oil. The oil is dissolved in EtOAc (20 mL), and treated with 4M HCI in dioxane (15 mL) at rt overnight. The white solid is collected via filtration, and washed with EtOAc. The solid is stirred in 20 mL of EtOAc and 25 mL of IN NaOH, the organic layer is separated, the aqueous layer is extracted with 20 mL EtOAc and organic layers are combined, washed with water (25 mL), brine (25 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The residue is purified by column chromatography on silica gel (9% MeOH:90% CH2Cl2: 1% TEA) to afford the title compound as a brown oil. LC-MS m/z (M+H): 263.09. 1H-NMR (400 MHz, CDCl3) δ: 7.18 (d, IH), 6.68 (d, IH), 3.8 (s, 3H), 3.6 (q, IH), 2.95 (d, IH), 2.74 (d, IH), 2.58 (m, IH), 2.26 (s, 3H), 2.17 (s, 3H), 2.00 (q, IH), 1.8 (d, IH), 1.7 (d, IH), 1.43-1.30 (br m, 5H), 1.20 (d, 3H).
Step 2. 2,5-Dichloro-iV-{ l-[l-(4-methoxy-2,3-dimethylphenyl)ethyl]piperidin-4-yl}nicotinaniide

2,5-Dichloronicotinoyl chloride is prepared from 2,5-dichloronicotinoyl acid (1.77 g, 9.22 mmol) by treatment with thionyl chloride (3.35 mL, 46.1 mmol, 5 eq.) at reflux for 18 h. The material is concentrated in vacuo, and used as is to acylate l-[l-(4-methoxy-2,3- dimethylphenyl)ethyl]piperidin-4-amine (900 mg, 3.43 mmol) under Schotten-Baumann conditions (ziO-propyl acetate, aqueous Na2CO3). Extractive work-up and purification by PTLC (2 mm silica gel plate) eluting with 2-3% MeOH (2M NHsVDCM, provides the title compound as a white, amorphous solid. 1H NMR: δ 8.40 (IH), 8.07 (I H), 7.21 (IH), 6.72 (IH), 6.35 (IH), 4.00 (IH), 3.81 (3H), 3.65 (I H), 2.96 (IH), 2.75 (I H), 2.28 (3H), 2.20 (2H), 2.18 (3H), 2.06 (2H);1.94 (IH), 1.56 (2H), 1.30 (3H), 1.27 (1H)I .2I (IH); LCMS: TR = 2.06 min, m/z 435.87 (M+ 1).
13. 5-CHLORO-2-(lSOPROPYLAMINO)-N-{ l -[I -(4-METHOXY-2,3 - DIMETHYLPHEΝYL)ETHYL]PIPERIDlΝ-4- YL)NICOTIN AMIDE

14. 5-CHLORO-2-[(3-ISOPROPOXYPROPYL)AMINO]-IV-{ L -[ L -(4-METHOXY-2,3- DIMETHYLPHENYL)ETHYL]PIPERIDΓN-4-YL}NICOTIN AMIDE

2s5-DichloiO-N-{ l -[] -(4-methoxy-2,3-dimethyIρhenyl)ethyl]piperidin-4-yl}nicotinamide (100 mg, 0.229 mmol) is dissolved in 5 mL xylenes, treated with 3-isopropoxypropan-l -amine (160 μL, 1.15 mmol, 5.0 eq.) and heated in a sealed tube in a 190 0C oil bath for 72 h. The solution is concentrated in vacuo and the residue purified by PTLC (2 mm silica gel plate) eluting with 3% MeOH (2M ΝH3)/DCM to provide the title compound as a brown oil. 1H NMR: δ 8.12 (I H), 8.00 (IH), 7.44 (IH), 7.21 (IH), 6.72 (IH), 5.82 (IH), 3.88 (I H), 3.81 (3H), 3.65 (IH), 3.5 (5H), 2.99 (IH), 2.76 (IH), 2.28 (3H), 2.18 (3H), 2.14 (2H), 2.00 (IH), 1.89 (3H), 1.49 (2H), 1.30 (IH), 1.17 (1 H), 1.15 (3H), 1.14 (3H); LCMS: TR = 2.30 min, m/z 517.19 (M+l).
15. 3 -CHLO RO-N- { I -[I -(4-MElΗOXY-2,3-DIMETHYLPHEΝYL)ETHYL]PIPERIDIΝ-4- YL}ISONICOTINAMIDE

3-Chloroisonicotinoyl chloride is prepared from 3-chloroisonicotinoyI acid (1.93 g, 12.25 mmol) by treatment with thionyl chloride (4.45 mL, 61.24 mmol, 5 eq.) at reflux for 18 h. The material is concentrated in vacuo, and used as is to acylate I-[l-(4-methoxy-2,3- dimethylpheny!)ethyl]ρiρeridin-4-amine (280 mg, 1.07 mmol) under Schotten-Baumarm conditions (/sø-propyl acetate, aqueous Na2CO3). Extractive work-up and purification by PTLC (2 mm silica gel plate) eluting with 5% MeOH (2M NHJXDCM provides the title compound as a tan solid. 1H NMR: 5 8.49 (IH), 7.62 (I H), 7.51 (I H), 7.22 (IH), 6.72 (IH)5 6.1 1 (IH), 3.95 (IH), 3.81 (3H),
3.68 (IH), 2.99 (IH), 2.78 (IH), 2.28 (3H), 2.20 (2H), 2.18 (3H), 2.02 (2H),1.94 (IH), 1.51 (2H), I .3O (3H),1.21 (IH); LCMS: TR - 2.01 min, m/z 401.89 (M+l).
16. 3-CHLORO-7V-{ l-[l-(4-METIIOXY-2,3-DlMETHYLPHENYL)ETHYL]PIPERIDIN-4-YL}BENZAMIDE

17. 3,5-DIMETHOXY-N-( ] -( ] -(4-METHOXY-2,3-DIMETHYLPHENYL)ETHYL)PIPERIDIN-4-
YL)BENZAM1DE

To a solution of l-(l-(4-methoxy-2,3-dimethylphenyl)ethyl)ρiρeridin-4-amine (0.105 g, 0.08 mmol, I eq.) in anhydrous toluene (0.8 mL) and TEA (0.34 mL, 0.24 mmol, 3 eq.) under N2 is added 3,5-dimethoxybenzoic acid (0.017 g, 0.096 mmol, 1.2 eq.). 2-Chloro-l,3- dimethylimidazolinium chloride (0.27 g, 0.160 mmol, 2 eq.) is then added portionwise to the mixture. The mixture is stirred at 50 0C for 3 h. The reaction is quenched by the addition of NaOH (IN, 0.5 mL) and extracted with EtOAc (2 x 0.5 mL). The organic layer is purified by SCX chromatography (0.5 g) to afford the title compound as a white solid. !H-NMR (400 MHz, CDCl3) δ: 6.85 (m, 2H), 6.72 (d, I H), 6.56 (m, IH), 5.9 (broad s, IH), 3.9 (m, IH), 3.8 (broad s, 8H), 3.7 (m, I H), 3.0 (m, IH), 2.8 (s, IH), 2.3 (s, 3H), 2.2 (broad m, 2H), 2.15 (s, 3H), 2.05 (broad d, IH). 1.95 (broad d, IH), 1.55 (broad m, 4H), 1.3 (broad s, 3H). m/z 427.19 (M+l).
18. N-((R)- 1 - { 1 - [4-(2-METHOXY-ETHOXY)-2,3 -DIMETHYL-PHENYL] -ETHYL } -P YRROLIDIN-2- YLMETHYL)-6-TRIFLUOROMETHYL-NICOTIN AMIDE
Step 1 . ] -[4-(2-methoxy-ethoxy)-2,3-dimethyI-phenyl]-ethanone

0.430 mol, 1.02 eq.), K2CO3 (85 g, 0.625 mol, 1.5 eq.), and Kl (12 g) is heated in CH3CN (350 mL) in a sealed flask at 1 100C overnight. The reaction mixture is cooled to rt, filtered through a Celite® pad and concentrated under reduced pressure. The residue is dissolved in ether (200 mL), washed with IN NaOH (3 x 150 mL) and brine (1 x 150 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give the desired intermediate without further purification. 1H-NMR (400 MHz, CDCl3) δ: 7.019-7.058 (t, I H), 6.77-6.79 (d, IH), 6.70-6.72 (d, I H), 4.09-4.11 (dd, 2H), 3.76- 3.78 (dd, 2H), 3.46 (s, 3H), 2.27 (s, 3H), 2.17 (s, 3H). To a suspension of aluminum chloride (53 g, 0.400 mol) in anhydrous CH2Cl2 (300 mL) under N2 at 0 0C is added acetyl chloride (27.4 g, 0.35 moi) slowly via an addition funnel, followed by the above intermediate (68 g, 0.38 mol). After stirring for 60 min at 0 0C, the reaction mixture is poured onto 300g of ice cubes, and vigorously stirred as cone. HCI (5 mL) is added slowly. After 1 h stirring, the organic layer is isolated, washed with brine, and dried over NaiSCV Removal of the solvent under reduced pressure affords the title compound as an off white oil which becomes white crystalline after storage in a refrigerator overnight. 1H-NMR (300 MHz, CDCl3) & 7.49-7.51 (d, IH), 6.70-6.72 (d, IH), 4.13-4.16 (dd, 2H), 3.77-3.79 (dd, 2H), 3.46 (s, 3H)3 2.53 (s, 3H), 2.41 (s, 3H), 2.19 (s, 3H). LC-MS m/∑ (M+H): 223.
Step 2. ((R)-I -{ l-[4-(2-Methoxy-ethoxy)-2,3-dimethyl-phenyl]-ethyI}-pyrrolidin-2- ylmethyl)carbamicacid tert-butyl ester

A mixture of (R)-l-pyiτoϊidin-2-ylmethyl-carbamic acid tert-butyl ester (Astatech Inc., Bristol, PA) (2.5 g, 12.5 mmol), l-[4-(2-methoxy-ethoxy)-2,3-dimethyf-phenyl]-ethanone (2.93 g,
13.12 mmol) and Ti(OiPr)4 (7.44 ml, 25 mmol) is heated to 70 0C overnight under a nitrogen atmosphere. The resulting mixture is cooled to rt and diluted with EtOH (50 ml). The solution is cooled to 0 0C, and NaBH4 (756 mg) is added. The resulting mixture is stirred at 0 0C to rt for 12 h, and is then quenched with 6 ml 1.6 M NaOH. The reaction mixture is stirred at rt for another 30 min. The suspension is filtered through Celite® and the Celite® is washed with acetone. After removal of solvent, the residue is purified by flash column chromatography (Hex/EtOAc = 1/1) to give the title compound. LCMS: TR = 1.36 min, mJz 407.40 (M+l).
Step 3. C-((R)-1 -{ I -[4-(2-Methoxy-ethoxy)-2,3-dimethy]-pπenyl]-ethyi} -pyrrolidm-2-yl)- methyl amine

To ((R)-l-{I-[4-(2-methoxy-ethoxy)-2,3-dimethyl-phenyl]-ethyl}-pyrrolidin-2-yimethyl)- carbarn ic acid tert-butyl ester (4 g. 9.85 mmol) in EtOAc (20 ml) is added 19.7 ml 4M HCI/dioxane at 0 0C, and the mixture is stirred at O 0C to rt overnight. After removal of solvent, water (30 ml) is added and the mixture is extracted with ether (3 x 25 ml) to remove impurities. The aqueous phase is made basic (pH>10) with 10 N NaOH and extracted with CH2CI2 (6 x 25 ml). The combined organic layers are washed with brine, dried and concentrated to give crude product, which is used in the next step without further purification. LCMS: TR = 1.98 min, m/z 307.09 (M+l).
Step 4. N-((R)-l-{ l-[4-(2-Methoxy-ethoxy)-2,3-dimethyl-phenyl]-ethyI}-ρyrroIidin-2-ylmethyl)-6- trifluoromelhyl-nicotinamide

To a mixture of C-((R)-l-{l-[4-(2-methoxy-ethoxy)-2,3-dimethyl-phenyI]-ethyl}- pyrrolidin-2-yl)-methylamine (31 mg, 0.1 mmol), 6-trifluoromethyI-nicotinic acid (0.2 M in DMF) (0.65 ml, 0.13 mmol) and BOP (66 mg, 0.15 mmol) in DMF (4 ml) is added TEA (0.25 mmol). After stirring at rt under a nitrogen atmosphere overnight, water (2 ml) is added and the mixture is extracted with EtOAc (3 x 8 ml). The combined organic layers are washed with brine, dried and concentrated. The crude product is then loaded onto a SCX column and the column is initially washed with 3 ml CH2Cl2/Me0H (3: 1) to remove impurities. The column is then washed with 3 ml EtOAc/MeOH/TEA (10:2: 1) and the title compound is obtained after removal of solvent. LCMS: TR = 2.06 and 2.10 min, m/z 480.25 (M+l). 1H NMR (CDCl3, 6 ppm): 8.90 (d, IH), 8.20-8.05 (dd, IH). 7.75 (m. I H), 7.23-7.15 (m, IH), 6.75-6.60 (m, IH), 4.05 (m, 2H), 3.78 (m, 2H), 3.40 (m. 3H). 3.39-2.45 (m, 6H), 2.25 (d, 3H), 2.10 (d, 3H). 2.0-1 .50 (in, 4H), 1.35 (m, 3H).
19. N-((S)- l -{ l -[4-(2-METMOXY-ETHOXY)-2,3-DIMETHYL-PHENYL]-ETHYL}-PYRROLrDIN-2- YLMETHYL)-O-TRIFLUOROMETHYL-NICOTINAMIDE
Step 1. (S)- l -Benzyl-pyiτolidine-2-carboxylic acid amide

Step 2. C-((S)-1 -Benzyl-pyrrolidin-2-yl)-methylamine

(S)-I -Benzyl-pyrrolidine-2-carboxyiic acid amide (40 g, 196.1 mmol) in 800 ml THF is added to a suspension of LiAlRi (37.2 g, 980.4 mmol) in 200 ml THF slowly. After addition, the mixture is refluxed under a nitrogen atmosphere overnight. After all starting material is consumed, the mixture is cooled to -40 0C and the reaction is quenched by carefully adding 38 ml H2O, 38 ml 15% NaOH, and 114 ml H2O sequentially. After the mixture is stirred at rt for 30 min, the solid is removed by filtering through Celite® and the Celite® is washed thoroughly with acetone. The solvent is removed under vacuum and crude title compound is obtained as a slightly yellow oil. LCMS: TR = 1.44 min, m/z 191 .09 (M+I).
Step 3. ((S)-l-Benzyl-pynOUdin-2-y!methyl)-carbamic acid tert-butyl ester

C-((S)-l-Benzyl-pyrrolidin-2-yl)-methylamine (21.3 g, 1 12.1 mmol) in 720 ml dioxane-H20 (2: 1) is cooled to 0 0C. Solid K2CO3 (30.9 g, 224.2 mmol) is then added, followed by (BOC)2O (29.3 g, 134.5 mmol). The resulting mixture is stirred at 0 0C to rt under a nitrogen atmosphere overnight. The organic solvent is removed and the residue is extracted with CH2Cl2 (4 x 300 ml). The combined organic layers are washed with H2O, dried and concentrated to give the title compound. LCMS: TR = 1.93 min, m/z 291.02 (M+ 1 ).
Step 4. (S)-l-Pyrrolidin-2-ylmethyl-carbamic acid tert-butyl ester

To ((S)-I -benzyl-pyrrolidin-2-ylmethyl)-carbamic acid tert-butyl ester (35 g) in 100 ml EtOH-AcOH (7:3) is added 20% Pd(OH)2/C (3.5 g). The resulting mixture is exposed to 50 psi H2 and heated to 55 °C on Parr Shaker overnight. After starting material is consumed (detected by
LCMS and TLC), the catalyst is filtered through Celite® and the Celite® is thoroughly washed with MeOH. The solvent is removed and the residue is taken into 150 ml H2O and made acidic with 6N HCL The mixture is extracted with ether (3 x 50 ml) to remove impurities. The aqueous phase is then made basic with 10 N NaOH and extracted with CH2CJ2 (4 x 100 ml). The combined organic layers are washed with H2O, dried and concentrated to give the title compound. LCMS: TR = 0.93 min, m/z 201.02 (M+l). !H NMR (CDCl3, δ ppm): 4.95 (s, IH), 3.20 (m, 2H), 2.95 (m, 3H), 1.95- 1.60 fm, 4H), 1.42 (s, 9H).
Step 5. ((S)-l-{ I-[4-(2-Methoxy-ethoxy)-2,3-dimethyI-phenyl]-ethyl}-ρyrrolidin-2-ylmethyl)- carbamic acid tert-butyl ester

A mixture of (S)-I -pyrrolidin^-ylmethyl-carbamic acid tert-butyl ester (3.86 g, 19.3 mmoi), l-[4-(2-methoxy-ethoxy)-2,3-dimethyl-phenyl]-ethanone (4.5 g. 20.27 mmol) and Ti(OiPr)4 (1 1.5 ml, 38.6 mmol) is heated to 70 0C under a nitrogen atmosphere overnight. The mixture is diluted with EtOH (80 ml) and cooled to 0 0C. NaBH4 (1.17 g, 30.9 mmol) is added and the resulting mixture is stirred at 0 0C to rt for 3 h. After all stalling material has been consumed, the mixture is cooled to 0 0C and 10 ml 6% NaOH is added to quench the reaction. After stirring at 0 0C for another 30 min, the solid is removed by filtering through Celite®. The solvent is removed under vacuum, and the residue is diluted with 50 ml H2O and made acidic with 6N HCI. The mixture is then extracted with ether (3 x 40 ml) to remove impurities. The aqueous phase is made basic with I O N NaOH and extracted with CH2CI2 (4 x 50 ml). The combined organic layers are washed with water (30 m!), dried and concentrated to give the title compound. LCMS: TR = 2.27 min, m/z 407.00 (M+l ). 1H NMR (CDCl3, δ ppm): 7.25-7.15 (m, IH), 6.75-6.65 (m, IH), 4.75 (s, IH), 4.10 (m, 2H), 3.95 (m, IH), 3.78 (m, 2H), 3.42 (s, 3H), 3.20-2.75 (m, 5H), 2.25 (s. 3H), 2.19 (s, 3H), 1.90-1.50 (m, 4H). 1.42 (m, 9H), 1.35 (m, 3H). Step 6. C-((S)-l-{ l -[4-(2-Methoxy-ethoxy)-2.3-dimethyl-phenyl]-ethyl}-pyrrolidin-2-yl)- methvlamine
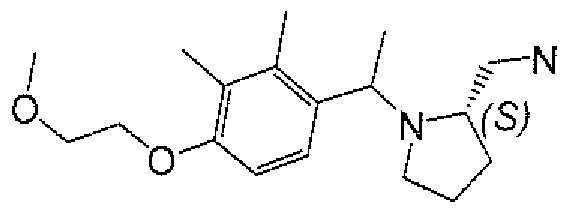
To ((S)-I -{ l -[4-(2-methoxy-ethoxy)-2,3-dimethyI-phenyl]-ethyl}-ρyrrolidin-2-ylmethyI)- carbamic acid tert-butyl ester (8 g. 19.7 mmol) in EtOAc (40 ml) is added 4M HCl/dioxane (40 ml) and the resulting mixture is stirred at rt overnight. After solvent is removed, the residue is dissolved in water (50 ml) and extracted with ether (3 x 35 ml) to remove impurities. The aqueous phase is then made basic with 10 N NaOH and extracted with CH2Cl2 (4 x 50 ml). The combined organic
layers are washed with water (30 ml), dried and concentrated to give the title compound. LCMS: TR = 1 .78 mins m/z 206.97 (M+l ). 1H NMR(CDCi3, δ ppm): 7.25-7.18 (m, 1 H), 6.70 (d, ϊ H), 4.10 (m, 2H), 3.95 (m, IH), 3.79 (ms 2H), 3.70 (s, 3H), 2.95-2.35 (m, 5H), 2.30 (s, 3H), 2.19 (s, 3H), 1.95- 1.40 (m, 4H), 1.35 (d, 3H).
Step 7. N-((S)-l-{ l-[4<2-Metlioxy-ethoxy)-2.3-dimethyl-ρhenyl]-ethyl}-pyrrolidin-2-yimethyl)-6- trifluoromethyl-nicotinamide

To a mixture of 6-trifluoromethyI-nicotinic acid (0.2 M in DMA; 0.24 ml) is added HBTU (0.2 M in DMA; 0.3 ml), followed by C-((S)-l-{ l-[4-(2-methoxy-ethoxy)-2,3-dimethyi-pheny!]- ethyl}-pyrrolidin-2-yl)-methylamine (0.2 M in toluene; 0.2 ml) and TEA (1 M in toluene; 0.15 ml) at rt. The resulting mixture is stirred at rt under a nitrogen atmosphere overnight. After the starting material is consumed, water (2 ml) is added and the mixture is extracted with EtOAc (3 x 5 ml). The combined organic layers are washed with brine, dried and concentrated, and the residue is loaded onto a SCX column. The column is washed with 5 ml CH2CVMeOH (3: 1) to remove impurities, and is then washed with 5 ml EtOAc-MeOH-TEA (10:2: 1). The title compound is obtained after removal of solvent. LCMS: TR = 2.31 min, m/z 480.04 (M+I).
20. 2-(3 ,4-DICHLORO-PΠENYL)-N-{(R)- 1-[4-(2-METHOXY-ETHOXY)-2,3 -DIMETHYL-BENZYL]- PYRROLIDIN-2- YLMETHYL}-ACETAMJDE
Step 1. {(R)-l-[4-(2-Methoxy-ethoxy)-2,3-dimethyl-benzyl]-pyrrolidin-2-ylmethyl}-carbamic acid fert-buty\ ester

To a mixture of (R)-l-pyrrolidin-2-ylmethyi-carbamic acid /erf-butyl ester (1.68 g, 8.4 mmol) and 4-(2-methoxy-ethoxy)-2,3-dimethyl-benzaldehyde (1.92 g, 9.24 mmoi) in 40 ml 1,2- dichloroelhane is added NaBH(OAc)3 (5.34 g, 25.2 mmol), followed by AcOH (0.24 ml, 4.2 mmol). The resulting mixture is stirred at 50 °C under a nitrogen atmosphere overnight. The reaction is quenched by adding 4 ml of 1 N NaOH at rt. After 30 min stirring at rt, the mixture is made acidic by 6 N HCl and extracted with ether (3 x 30 ml) to remove impurities. The aqueous phase is then made basic with 10 N NaOH and extracted with CH2Cl2 (4 x 40 ml). The combined organic layers are washed with water, dried and concentrated to give the title compound as a white solid. 1H NMR (CDCl3, δ ppm): 7.05 (d, IH), 6.65 (d, IH), 4.90 (m, IH)5 4.12 (t, 2H), 3.90 (d, IH), 3.75 (t, 2H), 3.45 (s, 3H), 3.35-2.60 (m, 5H), 2.25 (s, 3H), 2.20 (s, 3H)5 2.15-1.50 (m, 5H), 1.42 (s, 9H).
Step 2. C-{(R)-1 -[4-(2-Methoxy-ethoxy)-2,3-dimethyl-benzyl]-pyrroIidin-2-yl}-methylamine

To a mixture of { (R)-I -[4-(2 -methoxy-ethoxy)-2,3 -dimethyl-benzyl]-ρyrrolidin-2- ylmethyl} -carbarn ic acid
 ester (1.92 g, 4.9 mmol) in 50 ml EtOAc is added 7.3 ml 4 M HCl/dioxane. After overnight stirring at it under N2, water (40 mi) is added and the organic layer is separated. The aqueous layer is further extracted by ether (2 x 15 ml) before it is made basic with IO N NaOH solution. The aqueous phase is then extracted with CH2Cl2 (4 x 40 mi), dried and concentrated to give the title compound. LCMS: TR = 1.81 min, m/z 293.01 (M+ 1). !H NMR (CDCl3, δ ppm): 7.05 (d, IH), 6.65 (d, I H)5 4.10 (t, 2H), 3.95 (d, IH), 3.80 (t 2H), 3.45 (s, 3H), 3.20 (d, I H), 2.85-2.50 (m, 4H), 2.25 (s, 3H), 2.19 (s, 3H), 2.17 (m, IH), 1.95-1.60 (m, 4H).
ester (1.92 g, 4.9 mmol) in 50 ml EtOAc is added 7.3 ml 4 M HCl/dioxane. After overnight stirring at it under N2, water (40 mi) is added and the organic layer is separated. The aqueous layer is further extracted by ether (2 x 15 ml) before it is made basic with IO N NaOH solution. The aqueous phase is then extracted with CH2Cl2 (4 x 40 mi), dried and concentrated to give the title compound. LCMS: TR = 1.81 min, m/z 293.01 (M+ 1). !H NMR (CDCl3, δ ppm): 7.05 (d, IH), 6.65 (d, I H)5 4.10 (t, 2H), 3.95 (d, IH), 3.80 (t 2H), 3.45 (s, 3H), 3.20 (d, I H), 2.85-2.50 (m, 4H), 2.25 (s, 3H), 2.19 (s, 3H), 2.17 (m, IH), 1.95-1.60 (m, 4H).
Step 3. 2-(3,4-Dichloro-ρhenyl)-N-{(R)-l-[4-(2-methoxy-ethoxy)-2,3-dimethyl-benzyl]-pyrrolidin- 2-yImethyl}-acetamide

To a mixture of C-{(R)-l-[4-(2-methoxy-ethoxy)-2,3-dimethyl-benzyI]-pynOlidin-2-yl}- methylamine (0.2 M in DMF; 0.2 ml) and (3,4-dichIoiO-ρhenyl)acetic acid (0.2 M in DMF; 0.24 ml) is added BOP (0.2 M in DMF; 0.3 ml), followed by TEA (1.0 M in toluene; 0.4 ml). The mixture is stirred at rt under a nitrogen atmosphere overnight before it is quenched by H2O (2 ml), and extracted with EtOAc (2 x 2 ml). The combined organic layers are loaded onto a SCX column and washed with 3 ml CH2ClVMeOH (3: 1 ) to remove impurities. The SCX column is then washed with 3 ml EtOAc-MeOH-TEA (10:2: 1) and the title compound is obtained after solvent is removed. LCMS: TR = 1.20 min. m/z 478.18 (M+ 1).
21 . 2-(3,4-DLCHLORO-PHENYL)-N-{(S)-L -[4-(2-METHOXY-ETHOXY)-2,3-DIMETHYL-BENZYL]- PYRROIJDΓN-2-YLMETHYL}-ACETAMIDE
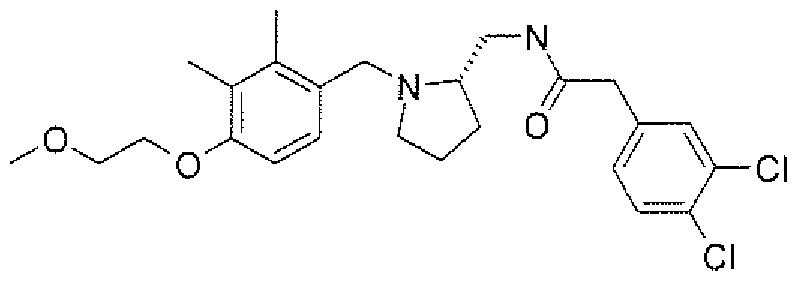
2-ylmethyl}-acetamide is prepared as described for Compound 20, with readily apparent modification of starting material. LCMS: TR = 1.29 min, m/z 479.12 (M+ 1).
22. N-{ l-[4-(4-DlMETHYLAMINO-BUTOXY)-2,3-DIMETHYL-BENZYL]-PIPERJDIN-2-YLMETHYL}-3,4- DIMETHYL-BENZAMIDE
Step 1. rac-[l-(4-Methoxy-2,3-dimethyl-benzyl)-piperidin-2-yImethyl]carbamic acid tert-butyl ester

A mixture of rac-piperidin-2-ylmethyl-carbamic acid tert-butyl ester (2.69 g, 12.6 mmol), A- methoxy-2,3-dimethyl-benzaldehyde (2.27 g, 13.8 mmol), NaBH(OAc)3 (8 g, 37.8 mmol) and HOAc (0.38 ml, 6.29 mmol) in 1,2-dichIoroethane (80 ml) is heated to 50 0C under a nitrogen atmosphere overnight. The reaction is quenched by adding 6 ml IN NaOH. The mixture is made acidic with 6 N HCl and extracted with ether (3 x 20 ml) to remove impurities. The aqueous phase is made basic with 10 N NaOH and extracted with CH2Cl2 (4 x 30 ml). The combined organic layers are washed with water (20 ml), dried and concentrated. The title compound is obtained after flash column chromatography (CH2Cl2/Me0H = 10/1). 1H NMR (CDCU, δ ppm): 7.05 (d, IH), 6.65 (d, I H). 4.95 (s, I H), 3.95 (d, I H), 3.80 (s, 3H), 3.40-3.25 (m, 2H), 3.23 (d, IH), 2.75 (m, IH), 2.42 (m, I H), 2.25 (s, 3H), 2.19 (s, 3H), 1.75-1.45 (m, 4H), 1.42 (s, 9H), 1.35 (m, 3H).
Step 2. rac-C-[l-(4-Methoxy-2,3-dimethyl-benzyl)-piperidin-2-yl]-methyiamine hydrochloride salt

To rac-[l-(4-methoxy-2,3-dimethyI-benzyl)-piperidin-2-ylmethyl]-carbamic acid /e/7-butyi ester (824 mg, 2.27 mmol) in 20 ml EtOAc is added 3.4 ml 4 M HCl/dioxane and the resulting mixture is stirred at rt under a nitrogen atmosphere overnight, and then at 50 0C for 3 h. After removal of solvent, the crude product is used for the next step without further purification.
Step 3. rac-N-[l -(4-Methoxy-2,3-dimethyl-benzyl)-piperidin-2-ylmethyl]-3.4-dimethyl-benzamide

To rac-C-[l-(4-methoxy-2,3-dimethyl-benzyl)-piperidin-2-yl]-methylamine hydrochloride salt (2.27 mmol) is added 3,4-dimethyl-benzoic acid (0.2 M in DMA; 13.6 ml), 2-chloro-1.3- dimethylimidazolinium chloride (0.2 M in CH3CN; 22.7 ml) and TEA (1 M in toluene; 1 1.4 ml).
After stirring at rt under a nitrogen atmosphere overnight 4 ml water is added and the mixture is extracted with EtOAc (3 x 30 ml). The combined organic layers are washed with brine, dried and concentrated. The residue is purified by column chromatography (CH2Cl2ZMeOH = 12/1 ) to give the title compound. LCMS: TR = 2.36 min, m/z 394.98 (M+l).
Step 4. rac-N-[l-(4-Hydroxy-2J3-dimelhyl-benzyl)-ρiρeridin-2-y1methyl]-3,4-dimethyl-benzamide

N-[l-(4-Methoxy-2,3-dimethyl-benzyl)-piperidin-2-ylmethyI]-3,4-dimethyI-benzamide (570 mg, 1.45 mrnol) in CH2Cl2 (20 ml) is cooled to -78 0C and BBr3 (I M in CH2Cl2; 7.9 mϊj is added dropwise. The resulting mixture is stirred under a nitrogen atmosphere at -78 0C to rt overnight.
After the starting material is consumed, the reaction mixture is carefully poured into ice-H2O, and solid Na2CO3 is added to make the solution basic (pH > 9). The organic phase is separated and the aqueous phase is extracted with CH2Cl2 (2 x 30 ml). The combined organic phases are washed with water (15 ml), dried and concentrated to give the title compound. LCMS: TR = 2.21 min, m/z 380.95 (M+ 1).
Step 5. rac-N-{l-[4-(4-Chloro-butoxy)-2,3-dimethyi-benzyl]-ρiρeridin-2-ylmethyl}-3,4-dimethyl- benzamide and N- { 1 -[4-(4-Iodo-butoxy)-2,3-dimethyI-benzyl]-piperidin-2-ylmethyl}-3,4-dimethyl- benzamide

Step 6. rac-N-{ l-[4-(4-Dimethylamino-butoxy)-2,3-dimethyl-benzyl]-piperidin-2-ylmethyl}-3a4- dimethyl-benzamide

23. 3,5-DIMETHOXY-N-((l -(l -(4-METHOXY-2,3-DIMETHYLPHENYL)ETHYL)PIPERIDIN-3- YL)MET!IYL)BE>JZAMIDE
Step 1. (l -(l-(4-methoxy-2,3-dimethylphenyl)ethy1)piperidin-3-yl)methanamine

A mixture of ] -(4-methoxy-2,3-dimethyiphenyI)ethanone (5.0 g, 0.028 mol. 1 eq.) and 3- (Boc-aminomethyl)piperidine (6.6 g, 0.031 mol, 1.1 eq.) and Ti(OiPr)4 (23.9 g, 0.084 mol, 3 eq.) is heated under N2 at 70 0C for 3 h. The reaction mixture is cooled to 0 0C, and anhydrous EtOH (150 mL) is added, followed by NaBH4 (1.55 g, 0.042 mol, 1.5 eq.) portionwise. The mixture is stirred at 0 0C for 1 h and then at rt overnight. The reaction is quenched by the addition of aqueous NaOH (1 N, 75 mL), and stirred at rt for 0.5 h. Insoluble materials are removed by filtration through Ceiite®, and the filter cake is washed with EtOAc. The filtrate and wash are combined and concentrated under reduced pressure. The residue is partitioned between water and EtOAc. The organic layer is washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure, and the residue is purified by flash chromatography (hexanes/EtOAc 1 : 1) to afford a yellow oil. The oil is dissolved in EtOA.c (30 mL), and treated with 4M HCI in dioxane (21 mL) at rt overnight. The white solid is collected via filtration, and washed with EtOAc. The solid is stirred in 100 mL of EtOAc and 50 mL of IN NaOH, the organic layer is separated, the aqueous layer is extracted with EtOAc and the organic layers are combined. Removal of the solvent affords the title compound as a pale yellow oil. m/z 111.1% (M+l).
Step 2. 3,5-dimethoxy-N-((l-(l-(4-methoxy-2,3-dimethylphenyl)ethyl)piperidin-3- yl)methyi)benzamide

To a solution of (l-(l-(4-methoxy-2,3-dimethylphenyl)ethyl)piρeridin-3-yl)methanamine
(0.2M in toluene, 0.10 mL, 0.02 mmol, 1 eq.) is added 3.5-dimethoxybenzoic acid (0.2 M in 95%
DMA and 5% TEA; 0.012 mL, 0.024 mmol, 1.2 eq.). A solution of BOP (0.2 M in dichloroethane;
0.15 mL, 0.04 mmol, 1.5 eq.) is then added to the mixture. The mixture is stirred at 50 0C overnight. The reaction is quenched by the addition of NaOH (IN, 0.5 mL) and extracted with
EtOAc (2 x 0,5 mL). The organic layer is purified by SCX chromatography (0.5 g). m/∑ 441.35 (M-H).
24. 3-CHLORO-N-(I -(I -(4~CYANO-2,3-DIMETHYLPHENYL)ETHYL)PΪPERIDΪN-4-YL)-4- FLU OROBENZAMIDE
Step 1 . ] -(l -(4-inethoxy-2.3-dimethy]pheny])ethyl)piperidin-4-amine
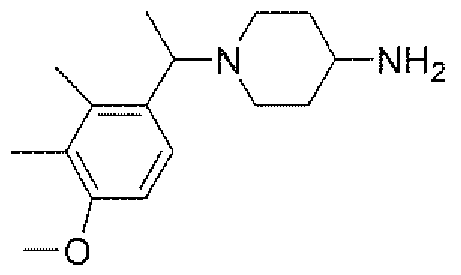
A mixture of l-(4-methoxy-2,3-dimethylphenyl)ethanone (4.45 g, 25.0 mmol) and 4-Boc- aminopiperidine (5 g, 25 mmol) in Ti(0iPr)4 (21.3 g, 75 mmol) under N2 is heated at 70 0C for 3 h. The reaction mixture is cooled to 0 0C, and anhydrous EtOH (150 mL) is added, followed by NaBH4 (1.43 g, 37.5 ramol) portionwise. The mixture is stirred at 0 0C for 1 h and then at rt overnight. The reaction is quenched by the addition of aq. NaOH (IN, 75 mL), and stirred at rt for 0.5 h. Insoluble materials are removed by filtration through Celite®, and the filter cake is washed with EtOAc. The filtrate and wash are combined and concentrated under reduced pressure. The residue is partitioned between water and EtOAc, the organic layer is washed with water and brine, dried over Na2SO4. and concentrated under reduced pressure, and the residue is purified by flash chromatography (hexane/EtOAc 1 : 1) to afford a yellow oil. The oil is dissolved in EtOAc (30 mL), and treated with 4 M HCl in dioxane (21 mL) at rt overnight. The white solid is collected via filtration, and washed with EtOAc. The solid is stirred in 100 mL of EtOAc and 50 mL of 1 N NaOH, the organic layer is separated, the aqueous layer is extracted with EtOAc, and the organic layers are combined. Removal of the solvent under reduced pressure gives the title compound as a yellow oil. 1H-NMR (400 MHz, CDCl3) δ: 7.22 (d, IH), 6.70 (d, I H), 3.80 (s, 3H), 3.62 (bs, IH), 2.9S (m, IH), 2.75 (m, IH), 2.62 (m, IH), 2.27 (s, 3H), 2.17 (s, 3H), 2.03 (m, 2H). 1.80 (m, I H), 1.70 (m, IH), 1.27-1.40 (m, 7H). LC-MS m/z (M+H): 263. Step 2. 3 -chloro-4-fluoro-N-( 1 -( I -(4-methoxy-2,3-dimethy tpheny!)ethy])piperidin-4-yl)benzamide

To a solution of l-(l-(4-methoxy-2,3-dimethylphenyl)ethyl)piperidin-4-amine (1.7 g, 6.49 mmol) in anhydrous CH2Cl2 (20 mL) under N2 at 0 0C is added TEA (1.36 mL, 9.74 mmol), followed by 3-chioro-4-fluoro-benzoyI chloride (1.32 g, 6.88 mmol). The reaction mixture is stirred at rt overnight. The reaction mixture is diluted with CH2Cl2, washed with IN NaOH and brine,
dried over Na2SO4, and concentrated under reduced pressure, and the residue is purified by flash chromatography (EtOAc) to afford the title compound as an off white solid. 1H-NMR (400 MHz, CDCl3) & 7.82 (d, IH), 7.61 (m, IH), 7.17-7.24 (m, 2H), 6.71 (d, IH), 5.89 (d, IH), 3.95 (m, IH), 3.81 (s, 3H), 3.68 (m, IH), 3.0 (m, IH), 2.80 (m, I H), 1.92-2.28 (m, 10H), 1.49 (m, 2H), 1.27 (m, 3H). LC-MS m/z (M+H): 419.25.
Step 3. 3-chIoro-4-f]uoro-N-( 1 -( 1 -(4-hydroxy-2,3-dimethyIphenyl)ethyl)piperidin-4-yl)benzamide

3-chIoro-4-fluoro-N-(l-(l-(4-methoxy-2,3-dimethyiphenyl)ethyl)piperidin-4-yl)benzamide (1.7 g, 4.0 mmol) is dissolved in CH2CI2 (20 mL), 1 M HCl in ether (8 mL) is added, and the solvent is removed under reduced pressure. The residue is dissolved in anhydrous CH2Cl2 (50 mL) under N2. The solution is cooled to -78 0C, and BBr3 (IM in CH2Ci2, 10 mL, 10 mmoi) is added dropwise. The mixture is allowed to warm to rt and is stirred overnight. The reaction mixture is cooled to 0 0C, and quenched with MeOH. The solvent is removed under reduced pressure. The residue is partitioned between EtOAc and 14 saturated NaHCθ3, and the aqueous layer is extracted with EtOAc. Organic layers are combined, washed with brine, dried over Na2SO4 and concentrated under reduced pressure to afford the title compound as a yellow solid. 1H-NMR (400 MHz, CDCl3) δ: 7.81 (d, IH), 7.62 (m, IH), 7.16-7.21 (m, 2H), 6.62 (d, IH), 5.87 (bs, IH), 3.95 (bs, IH), 3.69 (bs,lH), 3.04 (bs, IH), 2.80 (bs, IH), 1.91-2.27 (m, 10H), 1.56 (bs, 2H), 1.20-1.32 (m, 4H). LC-MS m/z (M+H): 405.24. Step 4. 3-chloro-N-(l -(I -(4-cyano-2,3-dimethylphenyl)ethyl)piperidin-4-yi)-4-fluorobenzarnide

To a solution of 3-chloro-4-fluoiO-N-(l-(l-(4-hydiOxy-2,3-dimethylphenyl)ethyl)piperidin- 4-yl)benzamide (0.4 g, 0.99 mmol) in anhydrous CH2Cl2 (7 mL) under N2 is added diisopropylethylamine (345 μL, 1.98 mmol), followed by N-phenyltrifhioromethanesuifonimide (0,71 g, 1.98 mmol). The mixture is stirred at rt for 72 h. The reaction mixture is diluted with CH2Cl2- washed with Vz saturated NaHCO3 and brine, dried over Na2SOjJ, and concentrated under reduced pressure. The residue is purified by PTLC (hexane/EtOAc/TEA: 50/50/1) to afford a yellow oil. To a solution of the oil (26.8 mg, 0.05 mmol) in anhydrous DMF (2 mL) in a tube is added Zn(CN)2 (29.4 mg, 0.25 mmol), Pd2(dba)3 (2.8 mg, 0.005 mmol) and DPPF (4.5 mg, 0.005 mmoi). After bubbling with argon for 3 min, the tube is sealed and heated at 80 0C with stirring
overnight. The reaction mixture is cooled to it, IN NaOH is added, and the mixture is extracted with EtOAc. The organic layer is washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by PTLC (hexane/EtOAc: 1/1) to afford the title compound as a yellow oil. 1H-NMR (400 MHz, CDCl3) & 7.82 (m, I H)1 7.64 (m, IHX 7.41-7.46 (m, 2H), 7.18 (t, IH), 6.07 (d, IH), 3.95 (M, I H)5 3.65 (q, IH), 3.05 <m, IH). 2.65 (m, I H), 2.50 (s, 3H), 2.30 (s, 3H), 2.05-2.38 (m, 2H), 1.90 (m, IH), 1.40-1.60 (m, 3H), 1 .25 (d, 3H). LC-MS m/z (M+H): 413.00.
25 AND 26. ALKYLATED DERIVATIVES OF 3-CMLORO-4-FLUORO-N-(3 -(1 -(4-HYDROXY-2,3- DIMETHYLPHENYL)ETHYL)PIPERIDIN-4-YL)BENZAMIDE 3-Chloro-N-(l-(l-(4-cyano-2,3-dimethylphenyl)ethyl)piperidin-4-yl)-4-fluoiObenzamide is alkylated with any of a variety of agents to generate compounds of the formula:

The following representative compounds are prepared in this manner:
25: R = < ^ N
A mixture of 3-chloro-N-(l-(l-(4-cyano-2,3-dimethylpheπyi)ethyl)ρiρeridin-4-yl)-4- fluorobenzamide (1 18 mg, 0.29 mmol), Cs2CO3 (190 mg, 0.58 mmoi) and 3-chloro-l-iodopropane (72 mg, 0.35 mmoi) in anhydrous DMF (2 mL) under N2 is heated at 40 0C overnight. The reaction mixture is cooled to rt, diluted with EtOAc, washed with water and brine, dried over Na2SO4 and concentrated under reduced pressure. The residue is purified by flash chromatography to afford a yellow solid. The solid is dissolved in anhydrous acetonitrile (5 mL), and treated with morpholine (182 μl) and cat. NaI and heated at 80 0C overnight. The reaction mixture is cooled to rt, and the solvent is removed under reduced pressure. The residue is partitioned between EtOAc and IN NaOH, and the EtOAc layer is washed with brine, dried over Na2Sθ4, and concentrated under reduced pressure. The residue is purified by PTLC (CH2Ci2/MeOH/NH,OH: 90/9/1) to afford Compound 25 as a yellow oil. 1H-NMR (400 MHz, CDCl3) δ: 7.82 (m, IH), 7.63 (m, IH), 7.16-7.23 (m, 2H), 6.70 (d, IH), 5.93 (d, IH), 3.94-4.01 (m, 3H), 3.69-3.74 (m, 5H), 3.01 (m, IH), 2.79 (m, I H), 2.55 (t, 2H), 2.47 (m} 4H), 2.17-2.28 (m, 8H), 1.91-2.03 (m, 4H), 1.54 (m, 2H), 1.30 (m, 3H). LC-MS m/z (M+H): 532.34.
26: R = -CH2-CH2-OH
To a solution of 3-ch!oro-N-(l-(l-(4-cyano-2,3-dimethy]pbenyl)ethyI)piperidin-4-yl)-4- fϊuorobenzamide (500 mg, 1.24 mmol) in anhydrous acetonitrile (12 mL) and DMF (3 mL) is added powdered KOH (139 mg, 2.48 mmol), followed by (2-bromoethyoxy)-/erf-butyldimethylsilane. The mixture is heated at 60 0C under N2 overnight. The mixture is cooled to rt, water is added, and the mixture is extracted with EtOAc. The organic layer is washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The residue is purified by flash chromatography (CH2C]2/MeOH: 95/5) to afford a yellow oil. The oil is dissolved in THF (15 mL) and cooled to 0 0C, and TBAF (IM in THF, 1.29 mL, 1.29 mmol) is added. After 30 min stirring at 0 0C, the reaction mixture is diluted with brine, and extracted with EtOAc. The organic layer is dried over Na2SO4 and concentrated under reduced pressure. The residue is purified by flash chromatography (CH2C]2MeOH: 97/3) to afford compound 26 as a yellow oil. LC-MS m/∑ (M+H): 448.10.
27. 3-CHLORO-N-(I -(I -(2,3-D1METHYLPYRJDIN-4-YL)ETHYL)PIPERIDIN-4-YL)-4-FLUOROBE1NZAMIDE Step 1. 4,5-Dimethyloxazo!e
Ox^N A mixture of 3-bromo-butan-2-one (50 g, 0.33 mol) and foπnamide (105 mL, 2.64 mo!) is heated for 5 h at 100 0C. The reaction mixture is cooled to rt and connected to a distillation head. The title compound is obtained as a clear liquid by distillation (collection starting at 30 0C at 25 mraHg). 1H-NMR (CDCl3): 57.70 (s, IH), 2.24 (s, 3H), 2.09 (s, 3H).
Step 2. 2,3-DimethyIisonicotinic acid ethyl ester

4,5-Dimethyloxazole (5 g, 51.5 mmol), EtOAc (1 1 ,2 mL, 103 mmol) and hydroquinone (catalyst, 50 mg) are heated in benzene (10 mL) at 85 0C for 16 h. The solvent is removed under reduced pressure and the residue is purified by silica gel flash chromatography eluting with hexanes- EtOAc (3:1) to afford the title compound as a yellow oil. 1H-NMR (CDCI3): £ 8.40 (d, I H), 7.39 (d, IH), 4.38 (q, 2H), 2.58 (s, 3H), 2.45 (s, 3H), 1.39 (t, 3H).
Step 3. (2,3-Dimethylpyridin-4-yl)-metlianol
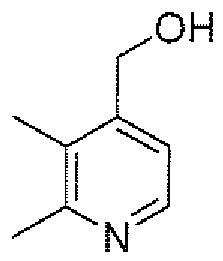
2,3-Dimethylisonicotinic acid ethyl ester (4.0 g, 22.3 mmol) is dissolved in anhydrous THF (100 mL) at 0 0C and treated with 1.5 equivalents of lithium aluminum hydride (33.5 mmol, IM
solution in THF) for 1 h. The flask is placed in an ice-water bath and the reaction is quenched by sequential addition of water (1.3 mL), 15% NaOH in water (1.3 mL), water (1.3 mL) and 13 g of MgSO4. The resulting suspension is stirred at rt for 30 min, and filtered through Celite®, and the filter cake is washed with 5% MeOH in DCM. The filtrate and washings are combined and concentrated under reduced pressure. The residue is purified by silicagel flash chromatography elitting with EtOAc to afford the title compound as an off-white solid. 1H-NMR (CDCl3): S 8.29 (d, IHX 7.24 (d, IH), 4.72 (s, 2H), 2.52 (s, 3H), 2.19 (s, 3H).
Step 4. 2,3-Dimethylpyridme-4-carbaldehyde

A mixture of 4A molecular sieves (2.1 g) in DCM (50 mL) is cooled to 0 0C under a nitrogen atmosphere. (2,3-Dimethylpyridin-4-y])-methanol (1.96 g, 14.3 mmoi) is added in one portion. 4-Methylmoφholine-N-oxide (3.35 g, 28.6 mmoi) is added and the resulting solution stirred for 10 min at 0 0C. Tetrapropylammonium perruthenate (266 mg, 0.76 mmoi) is added in small portions and the mixture is stirred at rt for 16 h. The reaction mixture is filtered through silica gel, eluting with hexanes-EtOAc (1 : 1). Removal of the solvents under reduced pressure produces the title compound as a clear oil. 1H-NMR (CDCl3): δ 10.38 (s, IH), 8.56 (d, IH), 7.45 (d, IH), 2.63 (s, 3H), 2.59 (s. 3H). Step 5. 3-chloro-4-Huoro-N-(ρiperidm-4-yf)beπzamide

To a suspension of l-Boc-4-aminopiperidine (2.2 g, 10.98 mmoi) in CH2Cl2 (20 mL) and sat. NaHCO3 (20 mL) at rt is added 3-chloro-4-fluoro-benzoyl chloride (3.2 g, 16.5 mmoi). After 1 h stirring, the organic layer is separated, washed with IN NaOH and brine, dried over Na2SOj, and concentrated under reduced pressure. The residue is dissolved in EtOAc (30 mL). and treated with 4M HCl in dioxane (30 mL) at rt overnight. The solid is collected via filtration and washed with EtOAc. The HCl salt is stirred in 100 mL of EtOAc and 50 mL of IN NaOH. The organic layer is washed with brine, dried over Na2SO4. and concentrated under reduced pressure to afford the title compound as a white solid. 1H-NMR (400 MHz, CDCl3): £ 7.84 (m, IH), 7.64 (m, IH)5 7.20 (t, I H), 5.93 (d, IH), 4.06 (m, I H), 3.12-3.15 (m, 2H)9 2.73-2.79 (m, 2H), 2.03-2.06 (m, 2H), 1.38-1.47 (m, 2H). LC-MS m/∑ (M+H): 257.06.
Step 6. 3-chloro-N-(l -( I -(2s3-dimethylpyridin-4-yl)ethyl)piperidin-4-yl)-4-fluoiObenzamide

To a solution of 3-chloiO-4-fluoro-N-(piperidin-4-yl)benzamide (12.8 mg, 0.05 mmol) in toluene- EtOH (0.6 ml, 2: 1) is added benzotriazole (6.8 rng, 0.058 mmol) and 2,3-Dimethylpyridinc- 4-carbaldehyde (7.4 mg, 0.055 mmoi). The reaction mixture is concentrated under reduced pressure at 40 0C. Toluene-EtOH (0.6 mL, 2: 1) is added and the evaporation is repeated, and this process is repeated once again. The resulting oil is dried under high vacuum for 2 h. The residue is dissolved in anhydrous THF (1 raL) and a solution of MeMgI (2M in THF, 0.075 mL) is added dropwise. The reaction mixture is stirred at rt for 20 min and then diluted with EtOAc, washed with IN NaOH, water and brine, dried over Na2SO4 and concentrated under vacuum. The residue is purified by silica gel PTLC. eluting with DCM-MeOH-ammonium hydroxide (90:9: 1) to yield the title compound as a colorless oil. 1H-NMR (400 MHz, CDCl3): ό~ 8.30 (d, IH), 7.82 (dd, I H). 7.65 (m. IH), 7.33 (d, IH), 7.19 (t, IH)5 5.98 (d, IH), 3.98 (m, IH). 3.69 (m, IH), 3.10 (m, IH), 2.73 (m, IH), 2.56 (s. 3H). 2.28 (s. 3H), 1.93-2.35 (m, 4H). 1.55-1.64 (m, 2H), 1.31 (d, 3H). LC-MS m/z: 390.27.
28. N- { 1 -[ 1 -(4-H YDROX Y-2,3 -DIMETH YLPHENYL)ETHYL]P YRROLIDIN-3 -YL} -3.5- DIMETHOXYBhNZAMIDE
Step 1. /e/7-Butyl (l-{ l-[4-(allyloxy)-2,3-dimethylpheπyi]ethyl}ρyrrolidin-3-yl)carbamate

A mixture of l-[4-(allyloxy)-2,3-dimethyIphenyl]ethanone (6.02 g, 29.5 mmol) and
(3R)-(+)-3-(ter/-butoxycarbonylamino)pyrrolidine(5 g, 26.8 mmol) in Ti(OiPr)4 (22.9 g, 80.5 mmol) under N2 is heated at 70 °C for 3 h. The reaction mixture is cooled to 0 0C, anhydrous EtOH (160 mL) is added, and followed by NaBH4 (1.53 g, 40.3 mmol) in small portions. The mixture is stirred at 0 0C for 0.5 h. The reaction is quenched by addition of aq. NaOH (IN, 160 mL). and stirred at rt for 0.5 h. Insoluble materials are removed by filtration through Ceiite*", and the filter cake is washed with EtOAc. The filtrate and wash are combined and concentrated under reduced pressure. The residue is partitioned between water and EtOAc, the organic layer is washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by flash silica gel chromatography (hexane/EtOAc: 1/1) to afford the title compound as a yellow oil. LC-MS m/z (M+l): 375.12.
Step 2. 1-{1 -[4-(allyloxy)-2,3-dimethylphenyl]ethyl}pyrrolidin-3-amine


To a solution of ] -{ l-[4-(ally!oxy)-2,3-dimethylphenyi]ethy!}pyiτolidin-3-amine (4.2 g, 12.1 mmol) in DMA (25 mL) is added TEA (6.75 mL, 48.6 mmol), 3,5-dimethoxybenzoic acid (2.43 g, 13.3 mmol) and BOP (8.05 g, 18.2 mmol). The mixture is stirred at it overnight. The reaction mixture is diluted with EtOAc, washed with IN NaOH, water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by flash silica gei chromatography (CH2CVMeOH: 9/1) to afford a yellow oil. The oil is dissolved in anhydrous CH2Cl2 (100 mL). Morpholine (1.16 mL, 13.4 mmol) is added. The solution is purged with nitrogen for 10 min, Pd(PPh3)4 (0.70 g, 0.61 mmol) is then added, and the reaction mixture is stirred under nitrogen for 2h. Solvent is removed under reduced pressure. The residue is diluted with EtOAc (15 mL), insoluble bright yellow catalyst is removed by filtration, and washed with EtOAc (2 x 15 mL). The filtrate and washes are combined, washed with Vi saturated NaHCO3 and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by silica gei chromatography (CH2Cl2ZMeOH: 95/5) to afford the title compound as a yellow solid.1HNMR (400 MHz, CDCl3) δ: 7.12 (m, I H), 6.84-6.88 (m, 2H), 6.50-6.60 (m, 2H), 4.62 (m, IH), 3.80 (s, 3H), 3.77 (s, 3H), 3.73 (m, I H), 3.55 (m, IH), 2.60-3.17 (m, 3H), 2.22-2.47 (m, 6H), 2.17 (s, 3H), 1.73 (m, IH), 1.31 -1.34 (m, 3H). LC-MS m/z (M+H): 399.13.
 DIMETHYLPHENYL)ETHYL]PYRROLIDINO -YL) -S3S-DIMETHOXYBENZAMIDE
DIMETHYLPHENYL)ETHYL]PYRROLIDINO -YL) -S3S-DIMETHOXYBENZAMIDE

To a suspension of NaH (60% in mineral oil, 60 mg, 1.51 mmol) in anhydrous DMF (1 mL) is added N-{ l-[l-(4-hydroxy-2,3-dimethylphenyl)ethyl]pyrrolidin-3-y]}-3,5-dimethoxybenzamide (400 mg, 1 mmol in 2 mL of DMF). The mixture is stirred at it for 30 min. (S)-(-)-glycidyl tosylate (344 mg, 1.51 mmol) is added. The mixture is stirred at rt overnight. The reaction mixture is poured into ιλ saturated NaHCO3 (5 mL), extracted with EtOAc. Organic phase is washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by flash chromatography (CH2Cl2/Me0H: 95/5) to afford the epoxide. To a solution of the epoxide (36.3 mg, 0.08 mmol) in iPrOH (1 mL) is added Me2NH (0.4 mL, 2M in THF). The mixture is heated at 80 0C overnight. The reaction mixture is concentrated under reduced pressure. The residue is dissolved in 2 mL of MeOH, loaded onto SCX cartridge (2 g of resin) which are pre- washed with MeOH (6 mL), and eluted with MeOH (2 x 6 mL) to remove non basic impurities, and EtOAc/MeOH/TEA (10/2/1, 6 mL) to afford the title compound as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.28 (m, I H), 6.87-6.91 (m, 2H), 6.72 (m, IH), 6.57 (m. IH), 6.52 (bs, IH), 4.61 (m, I H), 3.88-4.09 (m, 3H), 3.84 (s, 3H), 3.83 (s, 3H), 3.56 (m, IH), 2.22-3.10 (m, 17H), 2.18 (s, 1.5H), 2.17 (s, 1.5H), 1.73 (m, I H), 1.31-1.35 (m, 3H). LC-MS m/z (M+H): 500.31.
30. REL-N-{(3R)-l-[l-(4-{[(2R)-3-(DlMETHYLAMINO)-2-HYDROXYPROPYL]OXY}-2,3- DIMETHYLPHENYL)ETHYL]PYRROL]DIN-S-YL) O 5S-DIMETHOXYBENZAMIDE

The title compound is prepared as described above for Compound 29, using (R)-(+)-glycidyl tosylate instead of (S)-(-)-glycidyI tosylate. 1H NMR (400 MHz, CDCl3) δ: 12% (m, IH), 6.87-6.91 (m, 2H), 6.72 (m, I H), 6.57 (m, IH), 6.52 (bs, I H), 4.61 (m, I H), 3.88-4.09 (m, 3H)5 3.84 (s, 3H), 3.83 (s, 3H), 3.56 (m, IH), 2.22-3.10 (m, 17H), 2.18 (s, 1.5H), 2.17 (s, 1.5H), 1.73 (m, IH), 1.31- 1.35 (m, 3H). LC-MS m/z (M+H): 500.31.
31-35. ALKYLATED DERIVATIVES OF N- { 1 -[ 1 -(4-HYDROXY-2,3-
DIMETHYLPHENYL)ETHYL] P YRROLIDIN-3 -YL } -3 , 5 -DIMETHOX YBENZAMIDE
N-{ l-[l-(4-hydiOxy-2,3-dimethylρhenyI)ethyl]pynOlidiπ-3-yl}-3,5-dimethoxybenzamide is alkylated with any variety of agents to generate compounds of the formula:

The following representative compounds are prepared in this manner:
31 : R = -NMe2
To a suspension of N-{ l-[l-(4-hydroxy-2,3-dimethylphenyl)ethyl]pyrrolidin-3-yl}-3,5- dimethoxybenzamide (400 mg, 1 mmol) and Cs2CO3 (491.2 mg, 1.51 mmol) in DMF (5 mL) is added l -chloro-4-iodo-butane (147,6 μL, 1.21 mmol). The mixture is stirred at rt overnight. The reaction mixture is diluted with EtOAc, washed with water and brine, dried over Na2SO4 and concentrated under reduced pressure. The yellow oil residue is dissolved in 10 mL of anhydrous DMA as a stock solution. To 1 mL of this solution is added Cs2CO3 (48.9 mg, 0.15 mmol), 0.5 mL Of Me2NH (2M in THF7 1 .0 mmol) and NaI (cat.). The mixture is heated at 80 0C overnight, and is then diluted with EtOAc, washed with water and brine, dried over Na2SO4. and concentrated under reduced pressure. The residue is dissolved in 2 mL of MeOH, loaded onto SCX cartridge (2 g of resin) which are pre-washed with MeOH (6 mL), and eluted with MeOH (2 x 6 mL) to remove non basic impurities, and EtOAc/MeOH/TEA (10/2/1 , 6 mL) to afford the title compound as a yellow oil. 5H NMR (400 MHz, CDCl3) δ: 7.25 (m, IH), 6.87-6.91 (m, 2H), 6.69 (m, IH), 6.51-6.61 (m, 2H). 4.61 (m, IH), 3.91-3.96(m, 2H), 3.83 (s, 6H), 3.56 (m, IH), 2.29-3.10 (m, 8H), 2.28 (s, 1.5H), 2.27 (s, 1.5H), 2.23 (s, 3H), 2.22 (s, 3H). 2.17 (s, 1.5H), 2.16 (s, 1.5H), 1.62-1.85 (m, 4H). 1.31-1.35 (m, 3H). LC-MS m/z (M+H): 498.34.
32: R = -NMeEt
This compound is prepared as described for Compound 31, using methyl ethyl amine. 1H NMR (400 MHz, CDCl3) & 7.26 (m, IH), 6.88-6.92 (m, 2H), 6.69 (m, IH), 6.51 -6.61 (m, 2H), 4.61
(m, IH). 3.91-3.96(m} 2H), 3.83 (s, 3H), 3.82 (s, 3H), 3.56 (m, I H), 2.26-3.10 (m, 13H), 2.24 (s,
1.5H), 2.23 (s, 1.5H), 2.17 (s, 1.5H), 2.16 (s, 1.5H), 1.64-1.83 (m, 4H), 1.31-1.35 (m, 3H), 1.04-1.09
(m, 3H). LC-MS m/z (M+H): 512.26.
33: R = -NMe(CH2)2-OH This compound is prepared using 2-(methylamino)ethanol via the procedure described for
Compound 31, except that the crude product is purified by PTLC (CH2Cl2/MeOH/NH4OH: 90/9/1)
to afford the desired compound as a colorless oil. 1H NMR (400 MHz, CDCi3) S: 7.28 (m, IH), 6.87-6.90 (m, 2H), 6.69 (m, IH), 6.58 (m, IH)1 6.49 (bs, I H), 4.60 (m, IH), 3.92-3.95(m, 2H), 3.83 (S5 6H), 3.54-3.60 (m, 3H), 2.27-3.10 (m, 14H)5 2.26 (s, 1 .5H), 2.25 (s, 1.5H), 2.18 (s, 1.5H), 2.17 (s, 1.5H), 1.62-1.84 (m, 4H), 1.31-1.35 (m, 3H). LC-MS m/z (M+H): 528.24. 34: R = l-morphoiinyl
This compound (as a coiorless oil) is prepared using morphoiine via the procedure described for compound 33. !H NMR (400 MHz5 CDCl3) & 7.25 (m. IH), 6.86-6.89 (m, 2H), 6.69 (m, IH),
6.57 (m, I H), 6.46 (bs, IH), 4.60 (m, IH), 3.92-3.96 (m, 2H), 3.83 (s, 6H), 3.70-3.73 (m, 4H), 3.53
(m, IH), 2.22-3.10 (m, 15H), 2.17 (s, 1.5H), 2.16 (s, 1.5H), 1.67-1.84 (m, 4H), 1.30-1.34 (m, 3H). LC-MS m/z (M+H): 540.24.
35: R= 1-piperidinyi
This compound (as a colorless oil) is prepared using piperidine via the procedure described for compound 33. 1H NMR (400 MHz, CDCl3) δ: 12A (m, I H), 6.86-6.89 (m, 2H), 6.68 (m, IH),
6.57 (m, IH), 6.45 (m, IH), 4.60 (m, IH), 3.91-3.96 (m, 2H), 3.83 (s, 6H), 3.53 (m, IH), 2.22-3.08 (m, 15H), 2.17 (s, 3.5H), 2.16 (s, 1.5H), 1.67-1.84 (m, 4H), 1.54-1.61(m, 4H), 1.40-1.46 (m, 2H)9
1.29-1.34 (m, 3H). LC-MS m/z (M+H): 538.27.
36. 3-CHLORO-N-{ l-[l-(4-METHOXY-2, 3-DIMETHYLPHENYL)ETHYL]PYRROLJDJN-S-
YL} ISONICOTINAMIDE
Step 1. (3 R)-I-[I -(4-inethoxy-2,3-dijnethylphenyl)ethyl]ρyπOlidin-3-amine

This compound is prepared from l-(4-melhoxy-2,3-dimethylphenyi)ethanone (5.02 g, 28.2 mmol) and (3R)-(+)-3-(/er/-butoxycarbonylamino)pyrrolidine (5 g, 26.85 mmol) via the synthetic sequence described above for l-{ l-[4-(al!yloxy)-2,3-dimethylphenyl]ethyl}pyrrolidin-3-amine. The resulting HCl salt is partitioned between EtOAc and IN NaOH. The organic phase is washed with brine, dried over Na2SO4, and concentrated under reduced pressure to afford the title compound as a brown oil. !H-NMR (400 MHz, CDCl3 & 7.33 (d, IH), 6.72 (d, IH), 3.80 (s, 3H), 3.42-3.52 (m. 2H). 2.I 0-2.82 (m, I OH), 1.42-1.70 (m, 4H), 1.29-1.31 (m, 3H). LC-MS m/z (M+H): 249.12.
Step 2. 3-chloro-N-{ l-[l-(4-methoxy-2,3-dimethylphenyl)ethyl]pyrrolidin-3-yl}isonicotinamide

To a solution of (3R)-] -[I-(4-methoxy-2,3-dimethylphenyl)ethyl]ρyrrolidin-3-amine (0.06 mmol) in 0.3 mL of DMA is added 2-chloroisonicotinic acid (1 1.3 mg, 0.072 mmol), TEA (0.15 mmoi, IM in toluene) and DMC (0.12 mmol, 0.2 M in CH3CN). The mixture is stirred at 50 0C for 3 h. The reaction is cooled to rt, and is then diluted with EtOAc, washed with IN NaOH and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is dissolved in 2 mL of MeOH, loaded onto SCX cartridge (2 g of resin) which is pre-washed with MeOH (6 mL), and eluted with MeOH (2 x 6 mL) to remove non basic impurities, and EtOAc/MeOH/TEA (10/2/1 , 6 mL) to afford the title compound as a light brown oil. 1H NMR (400 MHz, CDCl3) & 8.51 (m, IH), 7.67 (s, 0.5 H), 7.63 (s, 0.5H), 7.53 (m, IH), 7.31 (m, I H), 6.60-6.74 (m, 2H), 4.61 (m, IH), 3.81 (s, 1.5H), 3.80 (s, 1.5H), 3.60 (m, I H), 3.16 (m, IH), 2.57-2.92 (m, 2H)5 2.22-2.46 (m, 6H), 2.17 (s, 1.5H), 2.16 (s, 1.5H), I .74 (m, I H), 1.33-1.37 (m, 3H). LC-MS m/z (M+H): 388.05.
37. 3-HYDROXY-5-METHOXY-N-{ l-[l-(4-METHOXY-2,3- DIMETHYLPHENYL)ETHYL]PYRROLIDIN-S-YL)BENZAM[DE

To a solution of (3R)-l -[l -(4-methoxy-2,3-dimethyiphenyl)ethyl]pyrrolidin-3-amine (1.24 g, 5 mmol) in DMA (20 mL) is added 3 -hydroxy- 5 -methoxy-ben zoic acid (0.882 g, 5.25 mmol), TEA (1.74 mL, 12.5 mmol) and BOP (3.32 g, 7.5 mmol). The mixture is stirred at rt overnight. The reaction mixture is diluted with EtOAc, washed with Vz saturated NaHCO3, water and brine, dried over Na3SOj, and concentrated under reduced pressure. The residue is purified by flash silica gel chromatography (EtOAc) to afford the title compound as an off white solid. 1H-NMR (400 MHz, CDCl3 δ: 7.60 (bs, IH), 7.36 (d, IH), 6.90-7.05 (m, 2H), 6.76 (m, IH), 6.35 (s, IH), 4.80 (m, I H), 3.70-3.94 (m, 7H), 3.42 (m, IH), 2.32-3.05 (m, 4H), 2.29 (s, 1.5H), 2.28 (s, 1.5H), 2.19 (s, 1.5H), 2.16 (s, I .5H), 1.56-1.86 (m, 2H), 1.52-1 ,56 (m, 3H). LC-MS m/z (M+H): 399.13.
38-42. ALKYLATED DERIVATIVES OF 3-HYDROXY-5-METHOXY-N- { 1-[1 -(4-METHOXY-2,3- DIMETHYLPHEN YL)ETH YL]P YRROLIDIN-3 -YL } BENZ AMIDE
3-hydroxy-5-methoxy-N-{ l-[l-(4-methoxy-2,3-dimethylphenyl)ethyl]pyrrolidin-3- yl}benzamide is alkylated with any variety of agents to generate compounds of the formula:

38: n=3, R = -NMe2
This compound is prepared from 3-hydroxy-5-methoxy-N-{ l-[l-(4-methoxy-2,3- dimethylphenyl)ethyl]pyrrolidin-3-yl}benzamide as described above for Compound 31. 1H-NMR (400 MHz, CDCl3) S: 7.30 (m, I H), 6.94 (m, IH), 6.90 (m, IH), 6.70-6.75 (m, 2H),. 6.57 (m, IH), 4.62 (m, I H), 4.02-4.07 (m, 2H), 3.83 (s, Ϊ .5H), 3.82 (s, 1.5H), 3.81 (s, 1.5H), 3.79 (s, 1.5H), 3.60 (m, I H), 2.20-3.15 (m, 15H), 2.17 (s, 1.5H), 2.16 (s, 1.5H), 1.64-2.02 (m, 4H), 1.33-1.36 (m, 3H). LC-MS m/z (M+H): 484.28.
39: n=3, R = I-morpholinyl This compound is prepared from 3-hydroxy-5-methoxy-N-{ l-[l-(4-methoxy-2,3- dimethylphenyl)ethyI]pyrro!idin-3-yl}benzamide as described above for Compound 34. 1H-NMR (400 MHz, CDCl3) & 7.30 (m, I H)5 6.86-6.90 (m, 2H), 6.72 (m, I H), 6.57 (m, IH), 6.46 (bs, I H)5 4.60 (m, IH), 4.04-4.07 (m, 2H), 3.83 (s, 3 H), 3.81 (s, 1.5H), 3.79 (s, 1.5H), 3.71-3.74 (m, 4H), 3.55 (m, IH), 2.21-3.10 (m. 13H), 2.17 (s, 1.5H), 2.16 (s, 1.5H), 1.94-2.01 (m, 2H), 1.61-1.81 (m, 2H), 1.32-1.35 (m, 3H), LC-MS m/z (M+H): 526.29.
40: n=4, R = -NMe2
This compound is prepared from 3-hydroxy-5~methoxy-N-{ l-[l-(4-rnethoxy-2,3- dimethylphenyl)ethyl]pyrrolidin-3-yI}benzamide as described above for Compound 31. 1H-NMR (400 MHz, CDCl3) S: 7.30 (m, IH), 6.85-6.89 (m, 2H), 6.71 (m, IH), 6.57 (m, I H), 6.47 (bs, IH), 4.62 (m, I H), 3.99-4.02 (m, 2H), 3.83 (s, 1.5H)5 3.82 (s, 1.5H), 3.81 (s, 1.5H), 3.79 (s. 1.5H), 3.54 (m, IH), 2.20-3.08 (m, 15H), 2.17 (s, 1.5H), 2.16 (s, 1.5H), 1.60-1.85 (m, 6H), 1.33-1.34 (m, 3H). LC-MS m/z (M+H): 498.21.
41 : n=4, R = 1 -morpholinyl
This compound is prepared from 3-hydroxy-5-methoxy-N-{ l-[l -(4-methoxy-2,3- dimethylphenyl)ethyl]pyrrolidin-3-yI}benzarnide as described above for compound 34. 1H-NMR
(400 MHz, CDCl3) & 7.30 (m, IH). 6.85-6.91 (m, 2H), 6.72 (m, IH), 6.56 (m, IH), 6.41 (bs, IH),
4.60 (m, IHX 3.99-4.02 (m, 2H), 3.83 (s, 3 H), 3.81 (s, 1.5H), 3.79 (s, 1.5H), 3.71-3.74 (m, 4H), 3.53 (m, IH), 2.21-3.08 (m, 13H), 2.17 (s, 1.5H), 2.16 (s, 1.5H)5 1,63-1.86 (m, 6H), 1.30-1.34 (m, 3HX LC-MS m/z (M+H): 540.21 .
42: n=4, R = -NMe(CH2)2-OH This compound is prepared from 3-hydroxy-5-methoxy-N-{ l-[l-(4-methoxy-253- dimethylphenyl)ethyl]pyrroIidin-3-yl}benzamide as described above for compound D-c. 1H-NMR (400 MHz, CDCI3) &. 12% (m, IH), 6.86-6.90 (m, 2H), 6.71 (m, IH), 6.56 (m, IH), 6.52 (bs, IH),
4.60 (m, IH), 3.99-4.03 (m, 2H), 3.83 (s, 1.5 H), 3.82 (s, 1.5H)1 3.81 (s, 1.5H), 3.79 (s, 1.5H)5 3.53-
3.61 (m, 3H), 2.20-3.08 (m, 15H), 2.17 (s, 1.5H), 2.16(s, 1.5H), 1.62-1.85 (m, 6H), 1.30-1.34 (m, 3H). LC-MS m/z (M+H): 528.21.
43-45. N-{ l-[l-(4-HYDROXY-2,3-DIMETHYLPHENYL)ETHYL]PIPERIDIN-3-YL}-3,5-
DiMETHOXYBENZAMiDE DERIVATIVES
The following analogs are made from (3R)-(+)-3-(/ert-butoxycarbonylamino)piperidine using the procedures described above. 43. 3,5-dimethoxy-N-((R)-l-(l-(4-methoxy-2,3-dimethylphenyl)ethyl)piperidin-3-yϊ)benzaniide

1H-NMR (400 MHz5 CDCi3) & 7.10 (d, IH), 6.78 (d, 2H), 6.63-6.69 (m, 2H), 6.55 (m, IH)5 4.15 (m, I H), 3.79-3.92 (m, 10H), 2.62-2.73 (m, 2H), 2.40-2.43 (m, 2H), 2.33 (s, 3H), 2.15 (s, 3H)5 1.50-1.85 (m, 4H), 1.29 (d, 3H). LC-MS m/z (M+H): 427.18.
44. N-((R)-l-(l-(4-(4-(dimethy]amino)butoxy)-2,3-dimethylphenyl)ethyl)piperidin-3-yl)-3,5- d i methoxy benzam ide

1H-NMR (400 MHz, CDCl3) & 7.14 (m. IH), 6.91 (m, 0.6H), 6.79 (d, 1.4H), 6.64-6.74 (m, 2H), 6.58 (t, 0.3H), 6.56 (t, 0.7H), 4.16 (m, IH), 3.79-3.96 (m, 8H), 3.67 (m, IH), 2.14-2.74 (m5 18H)5 1.51 -1.85 (m, 8H), 1.25-1.29 (m, 3H). LC-MS m/z (M+H): 512.15.
45. N-((R)- 1 -( 1 -C4-((R)-2-hydroxypropoxy)-2,3 -d imethyl phenyl)ethyl)piperidin-3 -yl)-3 , 5 - dimethoxybenzamide

3H-NMR (400 MHz. CDCl1) & 7.21 (d, 0.3H), 7.08 (d, 0.7H), 6.90 (d, 0.6H), 6.78 (d, 1.4H), 6.61- 6.76 (m, 2H). 6.58 (t, 0.3H), 6.56 (t, 0.7H), 4.14-4.22 (m, 2H), 3.67-3.92 (m, 10H), 2.13-2.69 (m, 10H), 1.50-1.84 (m, 4H), 1.22-1.29 (m, 6H). LC-MS m/z (M+H): 471.1 1.
46. N-{(3R)-l-[(l S)-l -(4-{[(2R)-2-HYDROXYPROPYL]OXY}-2,3- DIMETHYLPHENYL)ETHYL]PYRROLIDIN-3-YL}-3,5-DIMETHOXYBENZAMIDE
Step 1. Methyl (2R)-2-[(/1er/~butoxycarbonyI)amino]-4-oxobutanoate

A flask is charged with D-homo serine (1 g, 8.39 mmol), NaOH (336 mg, 8.39 mmol), water
(10 mL) and CH-,CN (10 mL). tert-Butyl dicarbonate is added, and the resulting solution is stirred at rt overnight. Solvents are removed under reduced pressure. The residue is dissolved in DMF (25 mL), iodomethane (10.1 mmol, 0.63 mL) is added, and the reaction is stirred at rt overnight. The reaction mixture is diluted with EtOAc (200 mL), washed with sat. NaHCO3 (3 x 50 mL), water (50 mL), O. I M Of KHSO4 (2 x 50 mL) and brine (50 mL), dried over Na2SO4, and concentrated under reduced pressure to afford a yellow oil. To a solution of the oil (0.41 g, 1.76 mmol) in anhydrous CH2Ci2 (10 mL) at 0 0C is added Dess-Martin periodinane (0.89 g, 2.1 1 mmol). After stirring at 0 °C for 2h, the reaction mixture is concentrated under reduced pressure to dryness. The residue is suspended in Et2O (60 mL). washed with aqueous NaHCO3/Na2S2O3 (100 mL of sat. NaHCO3 containing 25 g Of Na2S2O3), brine, dried over Na2SO4. and concentrated under reduced pressure to afford the title compound as a colorless oil. 1H NMR (400 MHz. CDCl3) δ: 9.73 (s, I H), 5.39 (bs. IH), 4.60 (m, I H), 3.75 (s. 3H), 2.96-3.1 1 (m, 2H), 1.42-1.46 (m. HH).
Step 2. Preparation of (R)- 1-[4-((S)-I -Amino-ethyl)-2,3-dimethyl-phenoxy]-ρropan-2-ol

D
A) (R )-Propylene oxide, K2CO3 B} TiCI4, CI2CHOCH3

Step 2A: Preparation of(R)-l-(2,3-Dimethyl-phenoxy)-propan-2-ol
To a mixture of 150.2 g 2,3-dimethylphenol (1.23 mol) and 75.0 g (R)-propylene oxide (1.29 mol) in 700 ml DMF is added 204 g K2CO3 (1.48 mol). The resulting mixture is heated to 120
0C for 5 h before it is cooled to rt. Part of the DMF is removed by evaporation and the residue is poured into 1.5 L water and extracted with ether (4 x 500 ml). The combined organic layer is washed with H2O (2 x 500 ml) and brine (200 ml). The aqueous layer is re-extracted with ether (150 ml x 2). The combined organic layer is dried and evaporated to afford the title compound. 1H NMR (CDCl3): 7.05 (t. I H), 6.80 (d, IH), 6.72 (d, IH), 4.25 (m, IH), 3.95 (dd, IH), 3.80 (dd, IH), 2.30 (s.
3H), 2.20 (s, 3H), 1.3O (d, 3H).
Step 2B: Preparation of4-((R)-2-Hydroxy-propoxy)-2,3-dimethyl-benzaldehyde
To 834 ml TiCl4 in CH2Ci2 (LO 3VI5 3 eq.) solution is added 37.4 ml Cl2CHOMe (1.5 eq) and the mixture is cooled to -78 0C. The (R)-I -(2,3 -Dimethyl-phenoxy)-propan-2-ol from Step A in 300 ml DCM is added dropwise at -78 °C and the resulting mixture is stirred at -78 0C to rt for overnight. After the reaction is complete, it is poured into cone. HCl (cooled at 0 0C) and the mixture is extracted with DCM (3 x 500 ml). The combined organic layer is washed with brine, dried and concentrated. After passing through a short silica gel pad, the mixture obtained is dissolved in 200 ml MeOH. TEA (5 ml) is added and the mixture is stirred at rt overnight. The mixture is concentrated and purified by column chromatography (Hex/EtOAc=3/l-l/l). 1H NMR (CDCl3): 10.20 (s. IH), 7.65 (d, IH), 6.80 (d, IH), 4.30 (m, IH), 4.00 (dd, IH), 3.90 (dd, IH), 2.60 (s, 3H), 2.20 (s, 3H). 1 .35 (d, 3H).
Step 2C: Preparation of 2-methyϊ-propane-2-sιdfmic acid l-[4-((R)-2-hydroxy-propoxy)-2,3- dimethyl-phenyl]-meth-(E)-ylideneamide To a mixture of 4-((R)-2-hydroxy-propoxy)-2,3-dimethyl-benzaldehyde (7.9 g. 38 mmol) in
120 ml DCM is added (R-)-(+)-2-methyl-2-propanesulfinamide (4.73 g, 39.1 mmol 1.03 eq), CuSO4 (15.2 g, 94.95 mmol, 2.5 eq) and PPTS (477 mg, 1.9 mmol, 0.05 eq), and the resulting mixture is stirred at rt overnight. After the reaction is complete, the mixture is filtered through Cefite®, washed
thoroughly with DCM. The filtrate is concentrated and purified by flash column chromatography (Hex/EtOAc - 2/1-1/1 ) to give the title compound. 1H NMR (CDCl3): 8.85 (s, I H), 7.82 (d. I H), 6.80 (d, I H), 4.25 (m, IH), 4.00 (dd, IH), 4.90 (dd, IH), 4.55 (s, 3H), 2.22 (s. 3H), 1.35 (d, 3H), 1.25 (s, 9H). Step 2D: Preparation of 2-methyϊ-propane-2-sulfinic acid {(S)-l-[4-((R)-2-hydroxy-propoxy)-2,3- dimethyl-phenyl]-ethyl}-amide
33.4 g of 2-methyl-propane-2-sulfinic acid l-[4-((R)-2-hydroxy-propoxy)-2,3-dimethyf- phenyI]-meth-(E)-ylideneamide (107.4 mrnol) in 850 ml dry DCM is cooled to 0 0C and 64.4 ml AiMe3 (2M in toluene) is added portionwise (exothermic). 179.4 ml MeMgCl (3M in THF) in 150 ml DCM is added dropwise at 0 0C. The resulting mixture is stirred at 0 0C to rt overnight. The reaction mixture is poured into ice and the mixture is filtered through Ceϋte®. The organic phase is collected and the aqueous phase is extracted with DCM (3 x 250 ml). The combined organic layer is dried and concentrated to give crude title compound. LCMS m/z: 328.09 (M+ 1).
Step 2E: Preparation of (R)-I -[4- ((S)-I -amino-ethyl)-2, 3-dimethyI-phenoxy]-propan-2-ol Crude 2-methyl-propane-2-sulfϊnic acid {(S)-l-[4-((R)-2-hydroxy-p]Opoxy)-2,3-dimethyl- phenyl]-ethyl} -amide (35g) is dissolved in 400 ml MeOH, and 100 ml 4N HCl/dioxane is added and the mixture is stirred at rt for 2 h. After ail starting material is consumed, the red solution is concentrated and the residue is taken into water (80 ml). The mixture is extracted with ether (2 x 80 ml) to remove impurities. The aqueous phase is then made basic with 1 ON NaOH and extracted with DCM (4 x 100 ml). The combined organic layer is washed with water (40 ml), dried and concentrated to give the title compound. Chiral HPLC analysis gives ratio of 16: 1 in favor of S- isomer. 1H NMR (CDCl3): 7.30 (d, IH), 6.75 (d, IH), 4.40 (q, I H), 4.20 (m, IH), 3.90 (dd, IH), 3.80 (dd, IH), 2.25 (s, 3H), 2.20 (s, 3H), 1.35 (d, 3H), 1.30 (d, 3H).
Step 3. Methyl (2R)-2-[(tø-/-butoxycarbonyl)amino]-4-{[(l S)-l-(4-{[(2R)-2-hydroxy propyljoxy}- 2.3-dimethylpheπyl)ethyl]amino}butanoate

OH
To a solution of methyl (2R)-2-[(/ert-butoxycarbonyl)amino]-4-oxobutanoate (0.38 g, 1.65 mmol) in anhydrous CHCl3 at 0 0C is added (R)-I -[4-((S)-I -amino-ethyl)-2,3-dimethyl-phenoxy]- propan-2-ol (0.368 g, 1.65 mmol). The solution is stirred at 0 0C for 30 min. NaBH(OAc)-. is added in small portion over 5 min. After stirring at 0 0C for 1.5 h, the reaction is quenched with addition of IN NaOH (10 niL). The mixture is extracted with CH2Cl2 (2 x), organic phases are combined, washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by flash chromatography (CH2Cl2ZMeOH: 9/1 ) to afford the title compound as a
pale yellow oil. 1H NMR (400 MHz, CDCl3) & 7.22 (d, IH), 6.73 (d, IH), 5.94 (bs, I H), 4.35 (bs, IH), 4.21 (m, IH), 3.98 (q, IH), 3.91 (dd, IH), 3.79 (m, I H), 3.71 (d, 2H), 3.47 (s, 3H), 2.51-2.62 (m, 2H), 2.23 (s, 3H), 2.19 (s, 3H), 3.94 (m, IH), 1.82 (m, IH), 1.59 (bs, 2H), 1.43 (s, 9H), 1.29 (d, 3H). 1.28 (d, 3H). LC-MS m/z (M+H): 439.05
Step 4. fe?-/-butyl {(3R)-l-[(l S)-l-(4-{[(2R)-2-hydroxyρropyI]oxy}-2,3-dimethylpheny])ethyl]-2- oxoρyrrolidin-3-yl} carbamate

OH
Methyl (2R)-2-[(/ert-butoxycarbonyl)amino]-4-{[(l S)-l-(4-{[(2R)-2-hydroxy propyl]oxy}-
2,3-dimethylρhenyl)ethyl]amino}butanoate (0.72 g, 1.64 mmol) is treated with aq. IN NaOH (8.2 mL) and EtOH (8.2 mL) at it overnight EtOH is removed under reduced pressure. The residue is diluted with water (5 mL), and pH is adjusted to 5-6. The white precipitates are collected, washed with water, and dried by evaporation with toluene under reduced pressure. The white solid is dissolved in DMA (20 mL), CH2Cl, (10 mL) and TEA (2 mL). BOP (0.82 g, 1.85 mmol) is added.
The mixture is stirred at 40 0C overnight. The reaction is cooled to rt, and CH2Cl2 is removed under reduced pressure. The residue is diluted with EtOAc, washed with IN NaOH, water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by silica gel chromatography (EtOAc/hexane: 1/2) to afford the title compound as a yellow solid. 1H NMR (400
MHz, CDCl3) & 7.13 (d, I H), 6.69 (d, IH), 5.51 (q, IH), 5.15 (bs, IH), 4.21 (m, IH), 4.1 1 (rn. IH).
3.92 (dd, IH), 3.79 (dd, IH)5 3.09 (t, IH), 2.48-2.59 (m, 2H), 2.18 (s, 3H), 2.1 1 (s, 3H), 1.58-1.78 (m. 2H). 1.50 (d, 3H), 1.44 (s. 9H), 1.30 (d, 3H). LC-MS m/z QA+23): 429.13.
Step 5. (3R)-3-amino-I-[(l S)-l-(4-{[(2R)-2-hydroxypropyI]oxy}-2.3 dimethylphenyl)ethyl] pyrrolidin-2-one

OH tert-butyl {(3R>l -[(l S>l -(4-{[(2R)-2-hydroxypropyl]oxy}-253-dimctliy]pheny])ethyl] -2-oxopyrrolidin-3-yl}carbamate (0.46 g, 1.13 mmol) is dissolved in 5 mL of EtOAc, and treated with 4M HCl in dioxane (2.3 mL) at rt overnight. Solvents are removed under reduced pressure.
The residue is partitioned between IN NaOH (10 mL) and EtOAc (20 mL). The white precipitate
(title compound) is collected. The filtrate is extracted with EtOAc/MeOH (95/5, 3 x). Organic phases are combined, washed with brine, dried over Na2SO4. and concentrated under reduced pressure to afford a yellow solid (additional title compound). The !H NMR of the yellow solid and
the white precipitate are identical. 1H NMR (400 MHz, CD3OD) δ: 7.20 (d, IH), 6.77 (d, IH), 5.41 (q, IH), 4.1 1 (m, I H)5 3.81-3.88 (tn, 2H), 3.49(dd, I H), 3.18 (m, I H), 2.52 (m, IH), 2.27 (in, IH), 2.18 (s, 3H), 2.08 (s, 3H), 1.65 (m, IH), 1.50 (d, 3H), 1.28 (d, 3H). LC-MS m/z (M+23): 328.91.
Step 6. (2R>ϊ-(4-{(lS)-l-[(3R)-3-ammopyrrolidin-l-yl3ethyl}-2,3-dimethylphenoxy)ρroρan -2-ol

OH
To a solution of (3R)-3-amino-l-[(l S)-l-(4-{[(2R)-2-hydroxypropyI]oxy}-2,3 dimethylphenyl)ethyl] pyrrolidin-2-one (0.3 g, 0.98 mmol) in anhydrous THF (30 mL) under N2 is added alane-dimethylethyJamine complex (0.5 M in toluene. 3.92 mL, 1.96 mmol). The mixture is stirred at rt overnight. The reaction is quenched with addition of aq. 2N NaOH (25 mL). After stirring at rt for 0.5 h, the organic layer is separated, and the aqueous layer is extracted with EtOAc (3 x 20 mL). The organic phases are combined, washed with brine, dried over Na2SO4, and concentrated under reduced pressure to afford the title compound as a yellow solid. 1H NMR (400 MHz, CDCl3) & 7.29 (d, IH)5 6.70 (d, I H), 4.21 (m, IH)5 3.92 (dd5 I H), 3.78 (dd, IH), 3.41 -3.52 (m, 2H), 2.54-2.68(m, 2H), 2.46 (m, IH), 2.34 (m, IH), 2.27 (s, 3H), 2.39 (s, 3H), 2.13 (m, IH), 1.50-1.60 (m, 4H), 1.29 (d, 3H), 1.28 (d, 3H). LC-MS m/z (M+l): 293.04.
Step ?. N-{(3R)-l-[(Ϊ S)-l-(4-{[(2R)-2-hydroxypiOpyl]oxy}-2,3-dimethylphenyl)ethyl]pyrrolidin -3 -y 1 } -3 , 5 -dimethoxy ben zam ide

To a solution of (2R)-l-(4-{(1 S)-l -[(3R)-3-aminopyπOlidin-l -yl]ethyl}-2,3- dimethylphenoxy)propan-2-oI (189 mg, 0.65 mmol) in DMA (5 mL) is added 3,5-dimethoxybenzoic acid (237 mg, 1.3 mmol), BOP (0.86 g, 1.94 mmol) and TEA (0.45 mL, 3.25 mmol). The mixture is stirred at rt overnight. The reaction mixture is diluted with EtOAc, washed with IN NaOH, water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is treated with IN NaOH (ImL) and EtOH (2 mL) at rt overnight. EtOH is removed under reduced pressure. The residue is diluted with EtOAc, washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by PTLC (CH2CI2/Me0H: 9/1) to afford the title compound as a yellow oil. 1H NMR (400 MHz, CDCl3) & 7.29 (d, IH), 6.90-6.91 (m, 2H), 6.70 (d, IH), 6.62 (bs, IH), 6.57 (t, I H), 4.65 (m, IH), 4.21 (m, IH), 3.91 (dd, IH), 3.77-3.83 (m, 7H), 3.56
(q, IH), 2.84 (m, IH), 2.75 (m, IH), 2.66 (m, IH), 2.42 (m, IH), 2.31 (m, IH), 2.28 (s, 3H), 2.24 (m, IH)5 2.19 (s, 3H), 1.68 (m, I H), 1.31 (d, 3H). 1.29 (d, 3H). LC-MS m/z (M+l): 457.06.
47. 3-OXA-9-AZA-BJCYCLO[3.3. l]NON-7-YL)-CARBAMIC ACID TERT-BUTYL ESTER Step 1. 2-(2,2-diethoxy-ethoxy)-l , 1 -diethoxy-ethane
EtCk /\Q/\ -0Et EtO OEt
2,2-Diethoxy-ethanol (100.8g. 752.2 mmol) is added to a mixture of NaH (60%) (31.9 g, 0.75 mol) and xylene (230 ml) at rt over 30 min and the resulting mixture is refluxed for Ih. After cooling to rt, 2-bromo-l,l-diethoxy-ethane (222.3 g, 1.13 mol) is slowly added and the mixture is refluxed overnight. After the reaction is cooled to rt, the precipitate is removed by filtration and the filtrate is concentrated under vacuum. The title compound is obtained after filtering through a silica gel plug with 300 mL of 1/1 Hexane/EtOAc. 1H NMR (CDCl3): 4.62 (t, 2H), 3.75 (m. 4H), 3.58 (q, 8H), 1 .21 (t, 12H).
Step 2. Preparation of 9-benzyl-3-oxa-9-aza-bicyclo[3.3.1]nonan-7-one

0.12N HCl (253 ml) and the mixture is stirred at rt overnight. Benzyl amine hydrochloride (86.6 g, 0 6 mo!) is then added, followed by NaOAc (22.4 g, 0.27 mol). Water (510 ml) and 3-oxo- pentanedioic acid (73.3 g, 0.5 mol) are added sequentially and the resulting mixture is stirred at rt overnight, followed by 1 h at 50 0C. The mixture is cooled to 0 0C and ION NaOH is used to raise the pH to 12. The mixture is extracted with EtOAc (4 x 300 ml) and the combined organic layer is washed with brine, dried and concentrated. The title compound is obtained after flash column chromatography (Hex/EtOAc=l/l). 1H NMR (CDCl3):7.45-7.25(m. 5H), 3.90(s, 2H). 3.82(d, 2H), 3.75(d, 2H), 3.20(d, 2H), 2.75(dd, 2H), 2.35(d, 2H).
Step 3. Preparation of 9-benzyl-3-oxa-9-aza-bicyclo[3.3.1]nonan-7-one oxime

To a mixture of 9-benzyl-3-oxa-9-aza-bicyclo[3.3.1]nonan-7-one (18.1 g, 78.4 mmol) in EtOH (120 ml) is added hydroxyamine hydrochloride (6 g. 86.2 mmol), followed by pyridine (7.6 ml, 94 mmol). The resulting mixture is refluxed for 2 h. The reaction is cooled to rt and 50 ml saturated Na2CO3 is added to quench the reaction. The mixture is extracted with DCM (4 x 100 ml) and the combined organic layer is washed with HjO. dried and concentrated to give the title compound. LC-MS m/z (M+H): 246.93, TR = 2.08 min.
Step 4. Preparation of 9-benzyi-3-oxa-9-aza-bicycIo[3.3.1]nαn-7-ylamine

A mixture of 9-benzyl-3-oxa-9-aza-bicycIo[3.3.1]nonan-7-one oxime {1.8 g, 7.3 mmol) in 1-pentanol (25 ml) is heated to reflux while sodium (2.0 g, 87.8 mmol) is added portionwise, and the mixture is refiuxed for 2h. The reaction mixture is made acidic (pH=l) with concentrated HCl at 0 0C and extracted with EtOAc (3 x 40 ml) to remove impurities. The aqueous phase is then made basic (pH>12) with ION NaOH and saturated with solid NaCl. The mixture is extracted with DCM (4 x 30 ml), and the combined organic layer is washed with H2O, dried and concentrated to give the title product. LC-MS m/z (M+H): 303.06, TR = 2.31min. Step 5. Preparation of (3-oxa-9-aza-bicyclo[3.3. l]non-7-yl)-carbamic acid tert-butyl ester

9-Benzyl-3-oxa-9-aza-bicyclo[3.3. I]non-7-ylamine (7.78 g, 33.5 mmol) is dissolved in DCM (100 ml), BoC2O (8.04 g, 36.9 mmol) is added and the mixture is stirred at rt overnight. The solvent is removed and the residue is titrated with 45 ml Hex/EtOAc (2: 1). The intermediate (9- benzyl-9-aza-bicyclo[3.3.1]non-3-y])-carbamic acid tert-butyl ester is collected as a solid after filtration. This obtained compound is dissolved in 250 ml EtOH and 10% Pd/C (1.9 g) is added under N2. The resulting mixture is exposed to 50 psi H2 on a Parr shaker for overnight. After ail starting material is consumed, the reaction mixture is filtered through Celite® and washed with EtOH. The combined EtOH wash is concentrated under vacuum to give the title product as a white solid. LC-MS m/z (M+H): 243.01, TR = 1.51 min.
48. 3-CHLORO-N-{9-[4-(4-DIMETHYLAMINO-BUTOXY)-2,3-DΪMHΠ IYL-BENZYL]-3-OXA-9-AZA- BLCYCLO[3.3. 1 ]NON-7-YL}-4-FLUORO-BENZAMIDE
Step 1. [9-(4-AI!y!oxy-2. 3-dimethyi-benzyl)-3-oxa-9-aza-bicyclo[3.3.3]non-7-yl]-carbamic acid /erf-butyl ester

To a stirred solution of (3-oxa-9-aza-bicyclo[3.3.1]non-7-yϊ)-carbamic acid tert-butyl ester (] , 1 g, 4.54 mmol) in 1 ,2-dichioroethane (20 ml) is added 4-allyloxy-2,3-dimethyl-benzaldehyde (0.95 g, 4.99 mmol), AcOH (0.26 ml, 4.54 mmol) and NaBH(OAc)3 (1.35 g) sequentially. The resulting mixture is allowed to stir at rt for 2 days, and is then quenched by the addition of 10 ml IN
KOH. After removal of solvent, the residue is treated with 100 ml 2N HCl and the mixture is extracted with ether (2 x 50 mi). The aqueous phase is then made basic (pH~12) with I ON NaOH and extracted with CH2Cl2 (3 x 100 ml). The combined CH2Cl2 layers are dried, concentrated and purified by flash coiurnn chromatography (Hex/EtOAc = 1/1 ) to give the title compound as a white solid. LCMS: TR = 1.40 min, m/z 417.23 (M+l).
Step 2. 9-(4-Allyloxy-2,3-dimethyl-benzyl)-3-oxa-9-aza-bicyclo[3.3. llnon-7-ylamine

To [9-(4-allyloxy-2,3-dimethyl-benzyl)-3-oxa-9-aza-bicyclo[3.3.1]non-7-yi]-carbamic acid tert-buty\ ester (0.48 g, 1.15 mmol) is added 4N HCl/dioxane (2 ml) and the resulting mixture is stirred for 2 h at it. After removal of solvent, the residue is made basic with 2N NaOH and extracted with CH2Cl2 (3 x 30 ml). The combined organic layers are dried and concentrated to give the title compound as a white powder. LCMS: TR = 1.07 min, m/z 335.17 (M+l).
Step 3. N-^^-Aliyloxy^^-dimethyl-benzyOo-oxa^^aza-bicyclopj. l Jnon^-yO^-chloro^- fluoro-benzamide
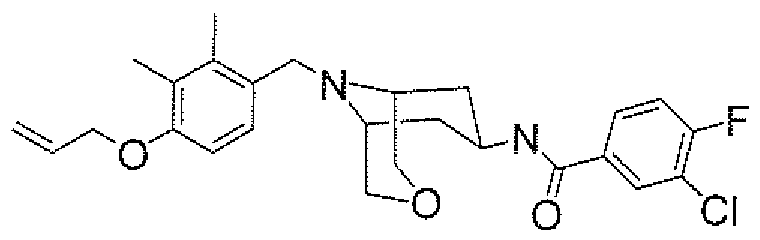
To a solution of 9^4-ai]yloxy-23-dimethyl4>enzyI)-3-oxa-9-aza4)icyclo[3.3.] ]non-7- ylamine (0.78 g. 2.47 mmol) in toluene (33 ml) is added a solution of 3-chloro-4-fiuoro-benzoic acid (0.52 g, 2.96 mmol) in CH3CN (15 ml), followed by TEA (0.86 ml, 6.16 mmol) and HBTU (1.40 g, 3.7 mmol). The mixture is stirred for 4 h at rt before it is quenched with 20 ml water. The solvent is removed under vacuum and the residue is treated with 100 ml saturated Na3CO3. The mixture is extracted with EtOAc (2 x 100 ml) and the combined organic layers arc washed with brine, dried and purified by flash column chromatography (Hex/EtOAc = 3/1) to afford the title compound. LCMS: TR = 1.44 min, m/z 473.20 (M+l).
Step 4. S-Chloro^-fluoiO-N-p^-hydroxy^^-dimethyl-benzyO-j-oxa^-aza-bicycio^.S.ljnon-?- yl]-benzamide

To a stirred solution of N-[9-(4-allyloxy-2,3-dimethyl-benzyl)-3-oxa-9-aza- bicyclop^.ljnon^-yll-j-chloro^-fluoro-benzamide (0.45 g, 0.95 mmol) in THF (5 ml) is added PPh3 (0.374 g, 1.43 mmol), morpholine (0.13 ml, 1.43 mmol) and Pd(OAc)2 (22 mg, 0.048 mmol) at
it. The mixture is stirred at rt for 2 days before it is diluted with CH2CI2 (80 ml) and washed with water (2 x 25 ml) and brine (25 ml). After drying, the organic layer is concentrated and purified by flash chromatography (CH2Cl2MeOH = 9/1) to give the title compound as a white solid. LCMS: TR = 1.30 min. m/z 433.10 (M+ 1).
Step 5. 3-Chloro-N-{9-[4-(4-chloro-butoxy)-2,3-dimethyl-benzyl]-3-oxa-9-aza-bicyclo[3.3.1]non- 7 -y 1 } -4 -flu oro-benzamid e

To a solution of 3-chloro-4-fluoro-N-[9-(4-hydroxy-2.3-dimethyl-benzyI)-3-oxa-9-aza- bicyclo[3.3.1]non-7-yi]-benzamide (0.28 g) in butanone (5 ml) is added Cs2CO3 (0.27 g) and 1- chloro~4-iodo-butane (0.47 ml). The resulting mixture is stirred at 100 0C for 16 h. The mixture is diluted with CH2Cl2 (50 ml) and washed with water (10 ml) and brine (10 ml). After diying over Na2SO4, the solvent is removed and the residue is purified by PTLC (CH2Cl2/Me0H = 20/1) to give a mixture of product with chloro- or iodo- at the terminal position. The mixture is used for the next step without further purification. Step 6. 3-Chloro-N~{9-[4-(4-dimethylamino-butoxy)-2,3-dimethyl-benzyl]-3-oxa-9-aza- bicyclo[3.3.1 jrton-7-y 1} -4-fluoro-benzaniide

A 25 ml sealed tube is charged with the mixture from step 5 (130 mg), CH^CN (30 ml), Ki
(10 mg). Cs2CO1 (0.12 g) and NHMe2 (0.1 ml). The resulting mixture is heated to 90 0C for 16 h. After removal of solvent, the residue is diluted with CH2Cl2 (50 ml), and washed with water (15 ml) and brine (15 ml). After drying over Na2SO4, the residue is purified by HPLC to give the title compound. LCMS: TR = 1.22 min. m/z 532.20 (M+ 1).
49. 3-CIlLORO-4-FLUORO-N-(l -(l-(4-(2-METHOXYETHOXY)-2,3- DIMEl HYLPHENYL)E1 HYL)PlPERIDIN-4- YL)BENZENESULFOM AMIDE Step 1. l-(l-(4-(2-methoxyethoxy)-2.3-dimethylphenyl)ethyl)piperidin-4-amine

A mixture of l -[4-(2-methoxyethoxy)-2.3-dimethylρheπyl]ethanone (5.54 g, 0.025 mo L 1 eq.) and 4-Boc-aminopiρeridine (5 g, 0.025 mol, 1 eq.) in Ti(OiPr)4 (14.2 g, 0.050 mol, 2 eq.) is
heated under N2 at 700C for 3 h. The reaction mixture is cooled to 0 0C, and anhydrous EtOH (75 mL) is added, followed by NaB H4 (1.42 g, 0.038 mol, 1.5 eq.) portionwise. The mixture is stirred at 0 0C for 1 h and then rt overnight. The reaction is quenched by addition of aq. NaOH (IN, 75 mL), and stirred at rt for 0.5 Si. Insoluble materials are removed by filtration through Celite®, and the filter cake is washed with EtOAc. The filtrate and wash are combined and concentrated under reduced pressure. The residue is partitioned between water and EtOAc. The organic layer is washed with water and brine, dried over Na2SO4, concentrated under reduced pressure, and the residue is purified by flash chromatography (hexanes/EtOAc 3: 1 ) to afford a yellow oil. The oil is dissolved in EtOAc (20 mL), and treated with 4M HCl in dioxane (20 mL) at rt overnight. The white solid is collected via filtration, and washed with EtOAc. The solid is stirred in 30 mL of EtOAc and 50 mL of IN NaOH, the organic layer is separated, the aqueous layer is extracted with 30 mL EtOAc and the organic layers are combined, washed with water (50 mL), brine (50 mL), dried over Na2SO^ filtered and concentrated under reduced pressure. Removal of the solvent affords the title compound as a pale yellow oil. LCMS: 307.20. 1H-NMR (400 MHz, CDCl3) δ: 7.18 (br d, I H), 6.68-70 (d, IH), 4.4 (br s, I H), 4.08-4.1 1 (dd, 2H), 3.75-3.78 (dd, 2H), 3.6 (br s, I H), 3.45 (s, 3H), 2.95 (br s, I H), 2.7 (br s, IH), 2.26 (s, 3H), 2.2 (s, 3H), 2.04-2.12 (br d, 2H), 1.86- 1.95 (br d, IH), 1.76-1.85 (br d, I H), 1.43 (s, 3H), 1.22-1.30 (br d, 2H).
Step 2. 3-Chloro-4-fluoiO-N-(l-(l-(4-(2-methoxyethoxy)-2,3-dimethylphenyl)ethyl)piperidin-4- yl)benzenesulfonamide

To a solution of l-(l-(4-(2-methoxyethoxy)-2,3-dimethylphenyl)ethyi)piperidin-4-amine
(0.030 g, 0.098 mmol, 1 eq.) in anhydrous CH3CN (0.3 mL) under N2 is added 3-chloro-4-fluoro- benzylsulfonyl chloride (0.025 g, 0.108 mmoi, Ll eq). The mixture is stirred at rt overnight. The reaction is quenched by the addition of NaOH (IN, 0.5 mL) and extracted with EtOAc (0.5 mL). The organic layer is purified by SCX chromatography (0.5 g) to afford the title compound as a white solid. 1H-NMR (400 MHz, CDCl3) δ: 7.95 (d, I H)5 7.74-7.75 (m, I H), 7.2-7.27 (t, I H), 7.1 (d, IH), 6.66 (d. IH), 4.07 (t, 2H)1 3.75 (t, 2H), 3.6 (q, IH), 3.45 (s, 3H), 3.12 (m, IH), 2.9 (q, IH), 2.83 (broad d, IH), 2.63 (broad d, IH), 2.23 (s, 3H), 2.18 (s, 3H), 1.95-2.05 (broad m, 2H), 1.75 (broad d, I H), 1.65 (broad d, I H), 1.35-1.5 (broad m, 2H), 1.23 (broad s, 3H). m/z 499.04 (M+I ).
50. 1 -(3,5-DIMETHOXYPHENYL)-3-( 1 -( 1 -(4-METHOX Y-2,3 -DIMETHYLPHENYL)ETHYL)PIPERIDIN^- YL)UREA

To a solution of l-(l-(4-methoxy-2,3-dimethyIphenyi)ethyl)piperidin-4-amine (0.008 g, 0.030 mmol, 1 eq.) in anhydrous toluene (0.3 mL) under N2 is added 3,5-dimethoxyρhenyl isocyaiiate (0.008 g, 0.045 mmol, 1.5 eq). The mixture is stirred at 60 0C for 6 h. The reaction is quenched by the addition of NaOH (IN, 0.5 mL) and extracted with EtOAc (0.5 mL). The organic layer is purified by SCX chromatography (0.5 g) to afford the title compound as a slightly yellow solid, m/∑ 442.39 (M+ 1).
51. N-(l-(4-(2-METHOXYETHOXY)-2,3-DIMETHYLBENZYL)PIPERIDlN-4-YL)-2-(6-(3,4- DICHLOROPHENYL)PYRIDlN-3-YL)ACETAMIDE
Step 1. 4-(2-methoxyethoxy)-2,3-dimethylbenzaldehyde

A mixture of 2,3-dimethylphenoϊ (36.65 g, 0.300 mol, 1 eq.), 2-chloroethyl methyl ether (28.35 g, 0.300 mol, 1 eq.), K2CO3 (53 g, 0.384 mol, 1.28 eq.), and KI (8.3 g, 0.050 mol, 0.17 eq.) is heated in CH3CN (350 mL) in a sealed flask at Ϊ OO 0C overnight. The reaction mixture is cooled to rt, filtered through a Celite® pad and concentrated under reduced pressure. The residue is dissolved in ether (200 mL), washed with IN NaOH (3 x 150 mL), brine (1 x 150 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give the desired intermediate without further purification. 1H-NMR (400 MHz, CDCt3) δ: 7.01-7.05 (t, IH), 6.77-6.79 (d, IH), 6.70-6.72 (d, IH), 4.08-4.1 1 (dd, 2H), 3.75-3.78 (dd, 2H), 3.46 (s, 3H), 2.26 (s, 3H), 2. Ϊ6 (s, 3H). This intermediate (21.2 g, 0.1 17 mol, 1 eq.) is dissolved in CH2Cl2 (150 mL) and is slowiy added to a stirring solution of 1.0M TiCl4 (234 mL, 0.234 mol, 2 eq.) and Cl2CHOCH3 (1 1.5 mL, 0.128 mol, 1.1 eq.) cooled to - 78°C. The reaction is allowed to slowly warm to rt and stirred overnight. The reaction is poured into ice (75 g) and cone. HCl (30 mL). The organic layer is separated, washed with water (100 mL) and brine (100 mL), dried over Na2SO.;, filtered and concentrated under reduced pressure. The residue is purified by column chromatography on silica gel (5% EtOAc: 95% Hexanes) to give the title compound. 1H-NMR (300 MHz, CDCl3) δ: 10.15 (s, IH), 7.63-7.66 (d, IH), 6.81-6.83 (d, IH), 4.17-4.20 (dd, 2H), 3.78-3.81 (dd, 2H), 3.46 (s, 3H), 2.59 (s, 3H), 2.21 (s, 3H).
Step 2. 1 -(4-(2-methoxyethoxy)~2,3-dimethylbenzyl)piperidin-4-amine

A mixture of 4-(2-methoxyethoxy)-2,3-dimethyIbenzaldehyde (5.0 g, 0.024 mol, 1 eq.) and
4-Boc-aminopiperidine (5.2 g, 0.026 mol, 1.1 eq.) in Ti(OiPr)4 (20.4 g, 0.072 mol, 3 eq.) is heated under N2 at 70 0C for 3 h. The reaction mixture is cooled to 0 0C, and anhydrous EtOH (150 mL) is added, followed by NaBH4 (1.37 g, 0.036 mol, 1.5 eq.) portionwise. The mixture is stirred at 0 0C for 1 h and then rt overnight. The reaction is quenched by addition of aq. NaOH (IN, 75 mL), and stirred at rt for 0.5 h. Insoluble materials are removed by filtration through Ceiite®, and the filter cake is washed with EtOAc. The filtrate and wash are combined and concentrated under reduced pressure. The residue is partitioned between water and EtOAc. The organic layer is washed with water and brine, dried over Na2SO4, concentrated under reduced pressure, and the residue is purified by flash chromatography (hexanes/EtOAc 3: 1) to afford a yellow oil. The oil is dissolved in EtOAc (40 mL), and treated with 4M HCl in dioxane (18.5 mL) at rt overnight. The white solid is collected via filtration, and washed with EtOAc. The solid is stirred in 30 mL of EtOAc and 50 mL of IN NaOH, the organic layer is separated, the aqueous layer is extracted with 30 mL EtOAc and the organic layers are combined, washed with water (50 mL), brine (50 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. Removal of the solvent affords the title compound as a pale yellow oil. Calculated for CnH28N2O2 292.22, found 293.15.
Step 3. N-(I -(4-(2-methoxyethoxy)-2,3-dimethyibenzyl)piperidin-4-yI)-2-(6-chloroρyridin-3- yl)acetamide

To a solution of l-(4-(2-methoxyethoxy)-2,3-dimethylbenzyl)piperidin-4-amine (1.55 g, 5.30 mmol, 1 eq.) in anhydrous DMF (20 mL) under N2 is added (2-chloropyridyl)-5-acetic acid (1.0 g, 5.83 mmol. 1.1 eq) and TEA (1.1 1 mL, 7.95 mmol, 1.5 eq.). BOP (2.81 g, 6.36 mmol, 1.2 eq.) is then added to the stirring mixture. The mixture is stirred at 50 0C overnight. The reaction is cooled to rt and diluted with EtOAc (30 mL). The organic layer is washed with NaOH (IN, 30 mL). H2O (30 mL) and brine (30 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting white precipitate is washed with Et2O and filtered to afford the title compound as a white solid. 1H-NMR (400 MHz, CDCl3) δ: 8.23 (d, IH), 7.62 (dd, IH), 7.3 (d, IH), 6.96 (d, IH). 6.64 (d, IH), 5.28 (d, IH), 4.1 (t, 2H), 3.75 (t, 2H), 3.45 (d, 4H), 3.35 (s, 2H), 2.75 (d, 2H), 2.23 (s. 3H), 2.18 (s, 3H), 2.08 (t, 2H), 1 .85 (d, 2H), 1.55 (broad s, 2H), 1.30-1.40 (broad m, 2H). m/∑ 446.16 (M+l).
Step 4. N-(l-(4-(2-methoxyethoxy)-2,3-dimethylbenzyl)piperidin-4-yl)-2-(6-(3J4- dichlorophenyl)pyridin-3-yl)acetamide

To a solution of N-(l-(4-(2-methoxyethoxy)-2,3-dimethylbenzyl)piperidin-4-yI)-2-(6- chioropyridin-3-yl)acetamide (0.009 g, 0.020 mmol, 1 eq.) in anhydrous dioxane (0.1 mL) under N2 is added a solution of 3,4-dichlorophenylboronic acid (0.008 g, 0.040 mmol, 2 eq) in anhydrous dioxane and a solution Of Na2CO.-; (IN, 0.05 mL, 0.050 mmol, 2.5 eq.) in H2O. The reaction is flushed with N2 for 20 min at rt. A solution of Pd(PPh3)4 (0.01M5 0.1 mL, 0.001 mmol, 0.05 eq.) in toluene is added. The mixture is stirred at 50 0C overnight. The reaction is quenched by the addition of NaOH (IN, 0.5 mL) and extracted with EtOAc (2 x 0.5 mL). The organic layer is purified by SCX chromatography (0.5 g). m/z 556.52 (M+ 1).
52. 2-CHLORO-N-( 1 -( I -(4-(2-MHTHOXYETHOXY)-2,3-DIMETHYLPHENYL)ETHYL)PIPERIDIN-4- YL)ISONICOTINAMiDE

This compound is prepared using the method described for Compound 31 , by reaction of I - (4-(2-methoxyelhoxy)-2,3-dimethylbenzyl)piperidin-4-amine with 2-chloroisonicotinic acid, m/z 446.1 1 (M+l).
53. N-(2,4-DlCHLOROBENZYL)- 1 -(I -(4-METHOXY-2,3-DIMRTΗYLPHENYL)ETHYL)PIPERJDIN-4-AMΪNE

To a solution of l-(l-(4-methoxy-2,3-dimethylpheiiy])ethyl)ρiρeridin-4-amine (0.2M, 0.1 mL, 0.02 mmol, 1 eq.) in anhydrous toluene and AcOH (0.2 M in toluene, 0.1 mL, 0.02 mmol, 1 eq.) is added a solution of 2,5-dichlorobenzaldehyde (0.2M, 0.15 mL, 0.03 mmol, 1.5 eq.) in toluene. The mixture is stirred at 50 0C overnight. A solution Of NaBH(OAc)3 (0.2M, 0.5 mL, 0.10 mmol, 5 eq.) is then added to the mixture. The mixture is stirred at rt overnight. The reaction is quenched by the addition of NaOH (IN, 0.5 mL) and extracted with EtOAc (2 x 0.5 mL). The organic layer is purified by SCX chromatography (0.5 g). m/z 421.24 (M+l).
54. 3-IIYDROXY-5-METHOXY-N-(l -(l -(4-METHOXY-2,3-DIMETHYLPHENYL)ETHYL)PIPERIDIN-4- YL)BENZAMIDE
Step 1. 3-hydroxy-5-methoxybenzoic acid

CH-,CN (100 niL) under N2 at 0 0C is added Cs2CO, (46.0 g, 0.140 mol, 1.02 eq). Jodomethane (6.9 mL, 0.1 10 mol, 0.8 eq.) is slowly added dropwise via addition funnel. The mixture is stirred at 0 0C for 1 h and then at rt overnight. The reaction is quenched by the addition of NaOH (IN, 300 mL) and extracted with CH2Ci2 (200 mL). The organic layer is separated and discarded. The aqueous layer is then acidified with HCl (IN, 300 mL) and extracted with CH2Cl2 (2 x 200 mL). The organic layers are combined, washed with brine, dried over Na2SO4, and concentrated under reduced pressure, and the residue is purified by flash chromatography (hexanes/EtOAc 4: 1 ) to afford an off- white solid, which is dissolved in NaOH (IN, 75 mL) and stirred at 50 0C overnight. The reaction is acidified to pH 5-6 with HCl (cone.) and extracted with diethyl ether (3 x 200 mL). The organic layers are combined, washed with brine, and dried over Na2SO4. Removal of the solvent under reduced pressure affords the title compound an as off-white solid. 1H-NMR (400 MHz, CD^OD) δ: 7.04 (broad s, 2H), 6.56 (s. IH), 3.27 (s, 3H). m/z 168.97 (M+l).
Step 2. 3-hydroxy-5-methoxy-N-( I -( 1 -(4-methoxy-2,3-dimcthyϊphenyl)ethyl)piperidin-4- yl)benzamide

To a solution of 3-hydroxy-5-methoxybenzoic acid (0.705 g, 0.004 mol, 1.1 eq.) and 1-(I- (4-methoxy-2,3-dimethylphenyI)ethyl)piperidin-4-amine (1.0 g, 0.003 mo!, 1.0 eq.) in anhydrous CH2CI2 (10 mL) and TEA (1.33 mL, 0.009 mol, 2.5 eq.) under N2 at 0 0C is added portionwise 2- chloro-l,3-dimethylimidazolinium chloride (0.97 g. 0.005 mol. 1.5 eq.). The mixture is stirred at 50 0C for 3 h. The reaction is quenched by the addition of NaOH (IN, 2 x 150 mL) and extracted with CH2Cl2 (15 mL), The organic layer is washed with brine, dried over Na2SO4, and concentrated under reduced pressure, and the residue is purified by flash chromatography (hexanes/EtOAc 3: 1) to afford the title compound as a yellow oil. 1H-NMR (400 MHz, CDCl3) S: 7.3 (d, IH), 6.7-6.9 (m. 3H), 6.5 (s, I H), 4.3-4.4 (m, IH), 3.9 (m, I H), 3.45 (broad d. IH). 3.25 (s, 3H). 3.15 (broad d, IH),
2.75 (broad s, 5H), 2.3-2.4 (broad m, 2H), 2.25 (s, 3H), 2.15 (s, 3H), 2.05 (s, 3H)5 1.85-1.95 (broad m 2H), 1.55 (d, 2Hλ m/z 413.25 (M+l).
55. 3-METHOXY-N-(l-(l-(4-METHOXY-2,3-DJMETHYLPHENYL)ETHYL)PlPERIDIN-4-YL)'5-(4- MORPHOLINOBUTOXY)BENZAMIDE

To a solution of 3-hydroxy-5-methoxy-N-(l-(l-(4-methoxy-2,3- dimethylphenyl)ethyl)piperidin-4-yi)benzamide (0.22 g, 0.525 mmol,, 1 eq.) in anhydrous DMF (2.0 mL) under N2 is added potassium t-butoxide (IM in THF, 0.78 inL, 0.786 mmol, 1.5 eq.). The mixture is stirred under N2 at it for 1 h, and then I-chloro-4-iodobutane (0.170 mg, 0.786 μmol) is added. The mixture is allowed to heat at 80 0C overnight. The reaction is cooled to rt, quenched with NaOH (IN, 10 mL) and extracted with 10 mL EtOAc. The organic layer is separated, washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by PTLC to afford the desired intermediate as an oil, which is dissolved in anhydrous CH3CN (1 mL) with K2CO3 (0.027 g, 0.199 mmol, 2.5 eq.). Morpholine (0.008 g, 0.096 mmol, 1.2 eq.) is added and the reaction is heated at 80 0C overnight. The reaction is allowed to cool to rt and is diluted with EtOAc (5 mL) and washed with NaOH (IN, 2 x 5 mL). The organic layer is separated, washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by PTLC to afford the title compound. 1H-NMR (400 MHz, CDCl3) δ: 7.22-7.24 (d, I H), 6.83 (d, 2H), 6.7 (d, I H),, 6.53-6.55 (m, IH), 5.90-5.92 (broad d, IH), 3.94-3.99 (t, 2H), 3.81 (s, 6H), 3.70-3.76 (m, 2H), 2.76-3.0 (broad m, 3H), 2.38-2.46 (m, 6H), 2.28 (s, 3H), 2.17-2.19 (m. 4H), 1.62-2.04 (broad m, 8H)5 1.41-1.50 (in, 2H), 1.23-1.27 (m, 5H). m/z 554.32 (M+l).
56. 3<4<DIMETHYLAMINO)BUTOXY)-5-METHOXY-N-(l-(H4-METHOXY-2,3- DIMETHYLPHENYL)ETHYL)PlPERIDIN-4-YL)BENZAMIDE

57. 3-(3-(DIMETHYLAMINO)PROPOXY)-5-METHOXY-N-(1 -(I -(4-MET110XY-2,3-
DiMETHYLPHEM YL)ETHYL)PΠΈRIDIN-4- YL)BENZAMIDE

This compound is prepared using the procedure described for Compound 43. with readily apparent modification of reactants. 1H-NMR (400 MHz, CDCI3) δ: 7.34 (d, IH), 6.87 (t, 2H), 6.74 (d, IH), 6.56 (t, IH), 3.95-4.18 (broad m, 2H)5 3.82 (s, 6H)7 3.2-3.4 (broad d, IH), 3.0-3.2 (m, 5H), 2.85-2.95 (broad d, IH), 2.8 (s, IH), 2.6 (t, IH), 2.38 (s, 3H), 2.27 (s, 3H), 2.18 (s, 3H), 1.92-2.09 (broad m, 3H), 1.64-1.72 (broad m, IH), 1.35-1.45 (m, 8H). m/z 498.22 (M+l).
58. 3-((R)-2-HYDROXYPROPOXY)-5-ME'H 10XY-N-(I -(I -(4-METHOX Y-2, 3- DIMETH YLPHENYL)ETHYL)PIPERIDlN-4-YL)BENZAMIDE

To solution of 3-hydroxγ-5-methoxy-N-( 1 -( 1 -(4-methoxy-2,3- dimethylphenyl)ethyl)piperidin-4-yl)benzamide (0.15 g, 0.364 mmol, 1 eq.) in anhydrous DMF (2.0 mL) under N2 is added potassium t-butoxide (IM in THF. 0.55 mL, 0.546 mmol, 1.5 eq.). The mixture is stirred under N2 at rt for 1 h, and then 2-{[;<?r/-butyl(dimethy!)silyI]oxy}propy] 4- methyl benzenesulfonate (0.18 g, 0.546 mmol. 1.5 eq.) is added. The mixture heated at 80 0C overnight. The reaction is cooled to rt, quenched with NaOH (IN, 10 mL) and extracted with EtOAc (2 x 10 mL). The organic layers are combined, washed with brine, dried over Na2SOj. and concentrated under reduced pressure. The residue is purified by PTLC to afford the desired intermediate as an oil, which is dissolved in anhydrous THF (1 mL) and cooled to 0 0C under N2. A solution of TBAF (IM in THF, 0.334 g, 0.334 mmol, 1.5 eq.) is slowly added and the reaction is allowed to slowly waπn to rt over 3 h. The reaction is quenched by the addition of NaOH (IN, 2 x 150 mL) and extracted with CH3Cl2 (15 mL). The organic layer is washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by PTLC to afford the title compound. 1H-NMR (400 MHz, CDCl3) δ: 7.24 (broad d, IH), 6.88 (broad s, 2 H), 6.7 (d. IH), 6.58 (t, IH), 6.59 (broad d, IH), 4.13 (m, 2H), 3.9 (m. I H), 3.8 (s, 6H), 3.7 (broad m, 3H), 3.45 (s,
2H), 3.28 (s, IH), 3.0 (broad d, IH), 2.8 (broad s, 2H), 2.1-2.3 (broad m, 7H), 2.0-2.1 (broad d, I H), 1.9-2.0 (broad d, I H), 1.5 (broad m, 2H)5 1.3 (broad d, 3H). m/z 471.13 (M+ 1).
59. 3-(2-HYDROXYETHOXY)-5-METHOXY-N-(] -(l-(4-METHOXY-2.3- DIMETHYLPHENYL)ETHYL)PIPERIDIN-4- YL)BENZAMIDE

To solution of 3-hydroxy-5-methoxy-N-(l -(I -(4-methoxy-2,3- dimethylρhenyϊ)ethyl)ρiρeridin-4-yl)benzamide (0,15 g, 0.364 mmol, I eq.) in anhydrous DMF (2.0 mL) under N2 is added potassium t-butoxide (IM in THF, 0.55 mL, 0.546 mmol, 1.5 eq.). The mixture is stirred under N2 at rt for 1 h, and then (2-bromoethoxy)(ter/-butyl)dimethyisilane (0.13 g, 0.546 mmol, 1.5 eq.) is added. The mixture is heated at 80 0C overnight. The reaction is cooled to rt and quenched with NaOH (IN, 10 mL) and extracted with EtOAc (2 x 10 mL). The organic layers are combined, washed with brine, dried over Na2SO.!, and concentrated under reduced pressure. The residue is purified by PTLC to afford the desired intermediate as an oil, which is dissolved in anhydrous THF (1 mL) and cooled to 0 0C under N2. A solution of TBAF (IM in THF, 0.189 g, 0.189 mmol, 1.5 eq.) is slowly added and the reaction is allowed to slowly warm to rt over 3 h. The reaction is quenched by the addition of NaOH (JN, 2 x 150 mL) and extracted with CH2Ci2 (15 mL). The organic layer is washed with brine, dried over Na2SO.!, and concentrated under reduced pressure. The residue is purified by PTLC to afford the title compound. 1H-NMR (400 MHz, CDCl3) δ: 7.2 (d, I H), 6.88 (broad s, 2 H), 6.7 (d, IH), 6.58 (t, IH), 4.1 (m, 2H), 3.9 (m, 2H), 3.8 (s, 6H), 3.7 (broad m, IH), 3.0 (broad d, IH), 2.8 (broad m, I H), 2.3 (s, 3b), 2.1 -2.25 (broad m, 4H), 1.8-2.1 (broad m, 2H). 1.5 (broad m, 2H), 1.3 (broad d. 2H), 0.9 (s, 5H). m/z 457.09 (M+l).
60. 3-METHOXY-N-(l -( l -(4-METHOXY-2,3-DIMETHYLPHENYL)ETHYL)PIPERIDIN-4-YL)-5-(2- METHOXYETIIOXY)BENZAMIDE

To solution of 3-hydroxy-5-methoxy-N-(l-(l-(4-methoxy-2,3- dimethylphenyI)ethyi)piperidin-4-yl)benzamide (0.1 g, 0.243 mmol, 1 eq.) in anhydrous DMF (2.0 mL) under N2 is added potassium tøv-butoxide (IM in THF, 0.364 mL, 0.364 mmol, 1.5 eq.). The mixture is stirred under N2 at rt for 1 h, and then 2-bromoethylmethylether (0.05 g, 0.364 mmol, 1.5
eq.) is added. The mixture is heated at 80 0C overnight. The reaction is cooled to rt, quenched with NaOH (IN, I O niL) and extracted with EtOAc (2 x 10 mL). The organic layers are combined, washed with brine, dried over Na2SO14, and concentrated under reduced pressure. The residue is purified by PTLC to afford the title compound. 1H-NMR (400 MHz, CDCi3) δ: 7.24 (broad d, IH), 6.88 (broad s, 2 H), 6.7 (d, I H)5 6.58 (t, IH)3 6.59 (broad d, I H), 4.13 (m, 2H), 3.9 (m9 IH), 3.8 (s, 6H), 3.7 (broad m, 3H), 3.45 (s, 2H), 3.28 (s, IH), 3.0 (broad d, IH), 2.8 (broad s, 2H), 2.1-2.3 (broad m, 7H), 2.0-2.1 (broad d, IH), 1.9-2.0 (broad d, IH), 1.5 (broad m, 2H), 1.3 (broad d, 3H). m/z 471.10 (M+l).
61. 2-(2-CHLORO -4 -FLUOROPHEN YL)-N-( 1 -( 1 -(4-METHOX Y-2 , 3 - DIMETHYLPHENYL)ETHYL)AZETIDIN-3-YL)ACET AMIDE
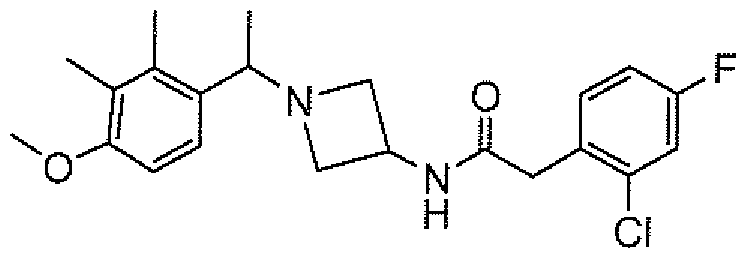
Step 1. l-(l-(4-methoxy-2,3-dimethylphenyl)ethyl)azetidin-3-arnine


To a solution of l-(]-(4-methoxy-2,3-dimethylpheπyl)ethyl)azetidin-3-amine (21.1 mg. 0.09 mmol) in DMA (2 niL) is added (2-chloro-4-fluoro-ρhenyl)acetic acid (0.099 mmol, 0.2 M solution in toluene/5% TEA) and BOP (59.7 mg, 0. Ϊ35 mmol). The mixture is stirred at rt overnight. The reaction mixture is diluted with EtOAc, washed with IN NaOH, water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by PTLC (EtOAc) to afford the title compound as a yellow oil. 1H-NMR (400 MHz, CDCl3) δ: 7.32 (m, IH), 7.22 (d, IH), 7.16 (m, IH), 6.99 (m, IH), 6.71 (d, IH), 6.02 (bs, IH), 4.48 (m, IH), 3.80 (s, 3H), 3.60-3.65 (m, 4H), 3.53 (t, IH), 2.98 (t, IH), 2.91 (t, IH)5 2.23 (s, 3H), 2.15 (s, 3H), 1 .15 (d, 3H). LC-MS m/z (M+H): 405.30.
62. 3-CHLORO-4-FLUORO-N-(l -(l -(4-METHOXY-2,3-DIMETHYLPHENYL)ETHYL)AZEΪIDIN-3- YL)BENZAMIDE

The title compound is prepared from l-(l-(4-methoxy-2,3-dimethy]phenyI)ethyI)azetidin-3- amiπe and 3-chloro-4-fluoro-benzoic acid essentially as described for Compound 51. 1H-NMR (400 MHz, CDCl3) & 7.90 (m, IH), 7.74 (m, IH), 7.28 (d, IH), 7.21 (t, IH), 6.90 (bs, IH), 6.74 (d, IH), 4.69 (m, IH), 3.80 (s, 3H), 3.61-3.70 (m, 3H), 3.18 (m, IH), 3.13 (m, IH), 2.54 (s, 3H), 2.17 (s, 3H), 1.20 (d, 3H). LC-MS m/z (M+H): 39] .31
63. 3-(2-FLuOROPHENYL)-N-(] -(l -(4-METHOXY-2,3-Dl'vϊETHYLPHENYL)ETHYL)AZETIDrN-3- YL)ACRYLAMIDE

The title compound is prepared from l-(l-(4-methoxy-2,3-dimethylphenyl)ethyl)azetidin-3- amine and 3-(2-fIuorophenyl)-arcylic acid essentially as described for Compound 51. 1H-NMR (400 MHz, CDCl3) & 7.68 (d, IH), 7.48 (m, IH), 7.27-7.33 (m, 2H), 7.06-7.16 (m, 2H), 6.72 (d, IH),
6.55 (d, I H), 6.28 (d, IH), 4.64 (m, IH), 3.80 (s, 3H), 3.54-3.65 (m, 3H), 2.98-3.04 (m, 2H), 2.24 (s, 3HX 2.16 (s, 3H), 1 .16 (s, 3H). LC-MS m/z (M+H): 383.36.
64. 3,5-DIMETHOXY-N-((l -(l -(4-METHOXY-2,3-DIMETHYLPHENYL)ETHYL)PYRROLIDΪN-3- YL)METHYL)BEMZAMIDE
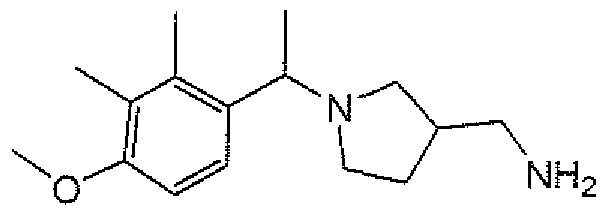


Step 1. (l-(l-(4-methoxy-2,3-dimethyiphenyl)ethyi)pyπ'θIidin-3-yl)methanamine
A mixture of l-(4-methoxy-2,3-dimethylphenyl)ethanone (1.78 g, 0.0 IO mol, 1.0 eq.) and pyrroIidϊn-3-ylmethyl-carbamic acid tert-bntyl ester (2.0 g, 0.010 mol, 1.0 eq.) in Ti(OiPr)4 (8.53 g, 0.030 mol, 3.0 eq.) is heated under N2 at 70 0C for 3 h. The reaction mixture is cooled to 0 0C, and anhydrous EtOH (60 mL) is added, followed by NaBH4 (0.59 g, 0.015 mol, 1.5 eq.) portionwise. The mixture is stirred at 0 0C for 1 h and then at rt overnight. The reaction is poured into a flask containing 70 mL Na2CO3 (minimal THF to transfer). The reaction is stirred at rt for 1 h, after which time 80 mL of diethyl ether is added. The biphasic solution is allowed to stir vigorously at it for 30 min. The organic layer is decanted off the white precipitate. The white solid is washed an additional two times with 80 mL diethyl ether in the same manner. The organic layers are combined and washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure, and the residue is purified by flash chromatography (hexanes/EtOAc 1 : 1) to afford a yellow oil. The oil is dissolved in EtOAc (8 mL), and treated with 4M HCS in dioxane (7.5 mL) at rt overnight. The white solid is collected via filtration, and washed with EtOAc. The solid is stirred in 10 mL of EtOAc and 10 mL of IN NaOH, organic layer is separated, aqueous layer is extracted with EtOAc and organic layers are combined. Removal of the solvent affords the title compound as a paie yellow oil. Calculated for C16H26N2O 262.4; found 262.99.
Step 2. 3,5-dimethoxy-N-((l-(l-(4-methoxy-2.3-dimethylphenyl)ethyl)pyiτolidin-3- yl)methyl)benzamide To a solution of (l-(l-(4-methoxy-2,3-dimethyiphenyi)ethyl)pyrroHdin-3-yl)methanamine
(0.2M in toluene, 0.10 mL, 0.02 mmol, 1 eq.) is added 3,5-dimethoxybenzoic acid (0.2M in 95%
DMA and 5% TEA, 0.012 niL, 0.024 mmol, 1.2 eq.). A solution of DMC (0.2M in dichloroethane, 0.20 mL, 0.04 mraol, 2.0 eq.) is then added to the mixture. The mixture is stirred at 50 0C for 3 h. The reaction is quenched by addition of NaOH (IN, 0.5 mL) and extracted with EtOAc (2 x 0.5 mL). The organic layer is purified by SCX chromatography to yield the title compound. Calculated for C25H34N2O4 426.55; found 427.22.
65. 4-(l-{4-[(3-CHLORO-4-FLUOROBEKZOYL)AMINO]PIPERIDIN-l-YL}ETHYL)-N-(2- M ETHOXY ETHYL)-N, 3 -DIMETH YLBENZ AMIDE
Step 1. Methyl 4-acety 1-3 -methylbenzoate

Step 2. 4-[l-(4-/-?r/-Butoxycarbonylamino-piperidin-l-yl)-ethyl]-3-methyl-benzoic acid isopropyl ester Boc

A mixture of methyl 4-acetyl-3-methylbenzoate (Ig, 5.21 mmol) and 4-Boc- aminopiperidine (1.04 g, 5.21 mmol) in Ti(OiPr)4 (4.5 g, 15.62 mmol) under N2 is heated at 70 0C for 3 h. The reaction mixture is cooled to 0 0C, and anhydrous EtOH (50 mL) is added, followed by NaBH4 (0.3 g, 7.8 mmol). The mixture is stirred at 0 0C for 1 h and then rt overnight. The reaction is quenched by addition of sat. NaHCO3, and stirred at rt for 10 min. The mixture is diluted with EtOAc, filtered through Celite®, and the filter cake is washed with EtOAc. The filtrate and wash are combined and concentrated under reduced pressure. The residue is partitioned between water and EtOAc. the organic layer is washed with water and brine, dried over Na2SO4, concentrated under reduced pressure, and the residue is purified by flash chromatography (hexane/EtOAcl : !) to afford
the title compound as a yellow oil. 1H-NMR (400 MHz, CDCI3) & 7.79-7.84 (m, 2H), 7.50 (d, IH), 5.23 (m, IH), 4.42 (bs, IH), 3.57 (q, IH), 3.43 (bs, IH), 2.97 (m, IH), 2.62 (in, IH), 2.38 (s, 3H), 1.77-2.12 (m, 4H), 1.34-1.50 (in, 17H), 1.25 (d, 3H). LC-MS m/z (M+H): 405.21.
Step 3. 4-{ l-[4-(3-Chloro-4-fIuoro-benzoylamino)-piperidin-l-yl]-ethyl}-3-methyl-benzoic acid isopropyl ester

4-[l-(4-/ert-Butoxycarbony]amino-piperidin-l-yI)-ethyl]-3-methyl-benzoic acid isopropyl ester (1.26 g, 3.12 mmo!) is treated with 4 M HCl in dioxane (6.2 mL) and EtOAc (6.2 mL) at rt overnight. Solvents are removed under reduced pressure. The off white solid is washed with Et2O, dried under reduced pressure to afford an off white solid. To a solution of the solid in anhydrous CH2Cl2 (15 mL) at 0 0C, is added TEA (1.54 mL, 11.1 mmol), followed by 3-chloro-4-fiuoro- benzoyl chloride (0.584 g, 3.04 mmol). The mixture is stirred at rt overnight. The reaction mixture is concentrated under reduced pressure. The residue is partitioned between Vz saturated NaHCO3 and EtOAc. The organic layer is washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue is purified by flash silica gel chromatography (hexane/EtOAc: 3/2) to afford the title compound as a yellow solid. 1H-NMR (400 MHz, CDCl3) δ: 7.81-7.86 (m, 3H), 7.57-7.63 (m, 2H), 7.19 (t, IH), 5.89 (bs, IH), 5.24 (m, I H), 3.97 (m, IH), 3.66 (bs, IH), 3.09 (bs, IH), 2.71 (m, IH), 2.41 (s, 3H), 1.58-2.46 (m, 4H), 1.28-1.65 (m, U H)-LC-MS m/z (M): 461.18. Step 4. 4-{ l-[4-(3-Chloro-4-fluoiO-benzoylamino)-piperidin-l-yl]-ethyl}-3-metliyI-benzoic acid

4-{ l -[4-(3-ChloiO-4-fluoro-benzoylamino)-piperidin-l-yl]-ethyl}-3-methyl-benzoic acid isopropyl ester (1.17 g, 2.54 mrnol) is treated with IN NaOH (13 mL) and EtOH (33 mL) at rt overnight. The resulting white suspension is treated with additional IN NaOH (13 mL) and EtOH (13 mL) at rt overnight. EtOH is removed under reduced pressure to give a white suspension. EtOAc (30 mL) is added, and the pH of the solution is adjusted to 6. The resulting white solid is collected, washed with water and Et2O, and dried by co-evaporation with toluene under reduced pressure to afford the title compound as a white solid. 1H-NMR (400 MHz, CD3OD-CDCI3) δ: 7.58-
7.93 (m, 4H), 7.48 (d, IH), 7.20 (t, I H), 3.77-3.91 (m, 2H), 3.30 (m, IH)5 2.85 (m, IH), 2.38 (s, 3H), 2.15-2.32 (m, 2H), 1.58-2.01 (m, 4H), 1.37 (m, 3H)XC-MS m/z {M): 419.03.
Step 5. 4-(l -{4-[(3-chloro-4-fluorobenzoyl)ammo]piperidin-] -yl}ethyl)-N-(2-methoxyethyl)-N,3- dimethylbenzamide

To a solution of 4-{ l-[4-(3-chioro-4-fluoiO-benzoylamino)-piperidin-l -yl]-ethyl}-3-methyl- benzoic acid (20.9 mg, 0.05 mmol) in DMA (1 mL) is added raethoxyethyl-methylamine (0.0575 mmol), TEA (17.4 μL, 0.125 mmol) and BOP (33.2 mg, 0.075 mmol). After stirring at il overnight, the mixture is diluted with EtOAc, washed with IN NaOH and brine, dried over Na2SO4 and concentrated under reduced pressure. The residue is dissolved in CH2Cl2 (2 mL), loaded onto SCX cartridge (2 g. pre-washed with 5 mL of MeOH), eluted with MeOH (5 mL) to remove nonbasic impurities, and then eluted with EtOAc/MeOH/TEA (10/2/1) to afford the title compound as an off white solid. 1H-NMR (400 MHz, CDCI3) δ: 7.83 (m, IH), 7.64 (m, IH), 7.43 (d, IH), 7.14-7.22 (m, 3H), 6.01 (d, IH), 3.93 (m, I H)5 3.68 (bs, 2M), 3.60 (q, IH), 3.31-3.51 (m, 4H), 2.98-3.15 (m, 4H), 2.60-2.75 (m, 2H), 2.37 (s, 3H), 1.82-2.20 (m, 4H), 1.40-1.56 (m, 2H), 1.28 (d, 3H). LC-MS m/z (M): 490.22.
66. 3,5-DlMETHOXY-N-{ l -[l -(4-METHOXY~2,3-DΪMETHYLPHENYL)ETHYL] PYRROLID!N-3-YΪJ}-N- (3 -METI 10XYPROPYL)BEN2AM1DE
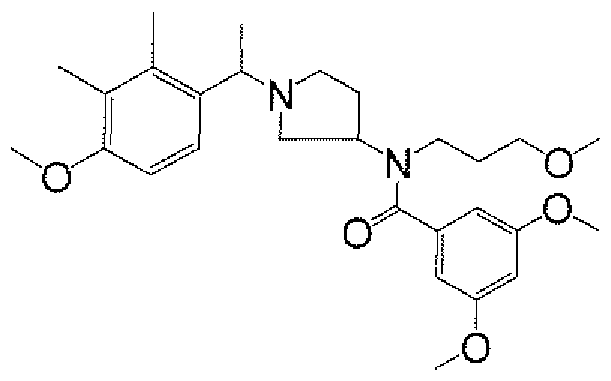
[l-(4-methoxy-2,3-dimethyl-phenyl)-ethyl]-pyn'olidin-3-yl}-benzamide (127.9mg, 0.31 mmol, 1 eq.) in DMF (2 mL). The reaction is allowed to stir at it for 30 min, Bromo-3-methoxypropane (95 mg, 0.62 mmol, 2 eq.) is added and the reaction is allowed to heat at 50 0C for 16 h. The reaction is quenched with IN NaOH (2 mL) and extracted with EtOAc (3 x 5 mL). The organic layers are combined, washed with IN NaOH (5 mL), water (5 mL) and brine (5 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting brown oil is purified by PTLC (95:5
CH2Cl2: MeOH) to afford the title compound as a yellow oil. LCMS: TR = 1.15 min. m/z 485.27 (M+l).
67. 3,5-DLMETHOXY-N- ( I -[I -(4-METHOXY-2,3-DLMETHYL-PHENYL)-ETHYL]-PΪPERIDIN-4-YL} -N- (2-MEΪHOXY-ETHYL)-BENZAMIDE

NaH (1.2 mg, 52.7 μmol, 1.5 eq.) is added as a solid to a solution of 3,5 -dim ethoxy -N-( I - [I-(4-methoxy-2,3-dimethyl-phenyl)-ethyI]-piperidin-4-yl}-benzamide (15 mg, 35.2 μmol, 1 eq.) in DMF (2 mL). The reaction is allowed to stir at rt for 30 min. Bromoethyl methyl ether (9.7 μL, 70.3 mmol, 2 eq.) is added and the reaction is allowed to heat at 50 0C for 16 h. The reaction is quenched with IN NaOH (2 mL) and extracted with EtOAc (3 x 5mL). The organic layers are combined, washed with IN NaOH (5 mL), water (5 mL) and brine (5 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting brown oil is purified by PTLC (3: 1 EtOAc: Hexanes) to afford the title compound. LCMS: TR - 1.01 min, m/z 485.29 (M+l ).
68. l-(2-METHOXYBENZYL)-3-{ 1 -[ 1 -(4-METHOXY-2,3-DIMETHYLPHENYL)ETHYL]PIPERIDIN-4-YL}- 1 ,3-BIS(2-METHOXYETHYL)UREA AND ϊ-(2-METHOXYBENZYL)-3-{ l-[l -(4-METHOXY-2,3- DIMETHYLPHENYL)ETHYL]PIPERIDIN-4- YL } - 1 -(2-METH0XYETHYL)UREA

NaH (8.9 mg, 423.5 μmol, 1 eq.) is added as a solid to a solution of l-(2-methoxy-benzyl)- 3-{ l-[I-(4-methoxy-2.3-dimethyl-phenyl)-ethyl]-piperidin-4-yl}-urea (180 mg, 423.5 μmol. 1 eq.) in DMF (2 mL). The reaction is allowed to stir at rt for 30 min. Bromoethyl methyl ether (40 μL, 423.5 μmol, 1 eq.) is added and the reaction is allowed to heat at 50 0C for 16 h. The reaction is quenched with IN NaOH (2 mL) and extracted with EtOAc (3 x 5 mL). The organic layers are combined, washed with IN NaOH (5 mL). water (5 mL) and brine (5 mL), dried over Na2SO4. filtered and concentrated under reduced pressure. The resulting brown oil is purified by PTLC (3: 1 EtOAc:hexanes) to afford l-(2-methoxybenzyl)-3-{ l-[l-(4-methoxy-2,3-dimethylphenyl)ethyl] piperidin-4-yl}-1.3-bis(2-methoxyethyl)urea (LCMS: TR = 1.16 min. m/z 542.35 (M+l)) and l-(2-
methoxybenzyl)-3-{ l-[l -(4-met!ioxy-2,3-dimethylphenyl)ethyl]piperidin-4-yI}-l-(2-methoxyethyl) urea (LCMS: TR= LOl min. m/z 484.30 (M+ 1)).
EXAMPLE 2. PREPARATION OF REPRESENTATIVE N-ALKYLATED AND ACYLATED N-ALKYLATED INTERMEDiATES N-Boc-4-piperidinone is reductively alkylated with any variety of agents to generate N- alkylated intermediates of the Formula:

The following representative intermediates are prepared in this manner: T-L tert-buty\ 4-[(2-methoxyethyS)amino]piperidine-I-carboxylate (n is 1 and R2o is methoxy):

A solution of N-Boc-4-piperidinone (19.93g, 100 mmol. 1 eq.) and 2-methoxyρroρylamine (9.47 mL, 110 mmol, I . I eq.) in 100 mL of DCE and acetic acid (2 niL) is treated with NaBH(OAc)3 (25.43 g, 120 mmol, 1.2 eq.) and stirred at 50 0C for 16 h. The reaction mixture is cooled to rt, diluted with 150 mL of IN NaOH and washed with 200 mL water, 200 mL brine, dried over Na2SO4, filtered and concentrated in vacuo to provide the title compound as a viscous yellow oil. 1H NMR: δ 4.01 <br, 2H), 3.47-3.50 (t, 2H), 3.35 (s, 3H), 2.73-2.81 (m, 4H), 2.55-2.60 (m, I H). 1 .79-1.83 (br d, 2H), 1.43 (s, 9H), 1.22-1.27 (m, 2H).
1-2. n is 1 and R20 is piperidine This intermediate is prepared using N-(2-aminoethyl)piperidinc via the procedure described above for intermediate I~l . LCMS: m/z 312 (M+ 1).
1-3. π is 1 and R2o is pyrrolidine
This intermediate is prepared using ] -(2-aminoethyl)pyiτolidine via the procedure described above for intermediate 1-1. LCMS: m/z 298 (M+ 1). 1-4. n is 1 and R20 is morpholine
This intermediate is prepared using 4-(2-aminoethyI)morpholine via the procedure described above for intermediate 1-1. LCMS: m/z 314 (M+ 1).
1-5. n is 1 and R20 is N(CH3),
This intermediate is prepared using N,N-dimelhy (ethylene diamine via the procedure described above for intermediate I- 1.
1-6. n is 1 and R20 is CH2N(CHa)2
This intermediate is prepared using N,N-dimethyl-l,3-propane diamine via the procedure described above for intermediate 1-1.
1-7. n is 1 and R20 is CH2OCH3 This intermediate is prepared using 3 -methoxypropy famine via the procedure described above for intermediate 1-1.
N-alkylated intermediates 1-1 to 1-7 are acylated with any variety of agents to generate acylated intermediates of the Formula:

The following representative intermediates are prepared in this manner:
1-8. ten 4-[(3,5-Dimethoxy-benzoyl)-(2-methoxy-ethyI)-amino3-piperidine-l-carboxylic acid tert- butyl ester

1-9. n is 1 and R2O is piperidine
This intermediate is prepared from 1-2, via the procedure described above for 1-8. LCMS: TR = 1.37 min m/z 476.19 (M-H). I- 10. n is 1 and R2o is pyrrolidine
This intermediate is prepared from 1-3, via the procedure described above for 1-8. LCMS: TR = 1.36 min /;?/;: 462.19 (M+ 1).
ϊ-1 1. n is I and R2o is morphoϋπe
This intermediate is prepared from the 1-4. via the procedure described above for 1-8. LCMS: TR = 1.35 min m/z 478.18 (M+ 1).
1-12. n is 1 and R20 is N(CH3), This intermediate is prepared from 1-5, via the procedure described above for 1-8. LCMS:
TR = 1.35 min m/z 436. ] 4 (M+ 1 ).
1-13. π is 1 and R20 is CH2N(CH3)2
This intermediate is prepared from 1-6, via the procedure described above for 1-8. LCMS: TR = 1.35 min m/z 450.16 (M+ 1). ϊ- 14. n is 1 and R20 is CH2OCH3
This intermediate is prepared from 1-7, via the procedure described above for 1-8. LCMS: TR = 1.55 min m/z 437.12 (M+l).
Acylated N-alkylated intermediates of intermediates 1-8 to 1-14 are deprotected to generate intermediates of the Formula:

The following representative deprotected intermediates are prepared in this manner:
1-15. 3,5-Dimethoxy-N-(2-methoxy-ethyl)-N-piperidin-4-yl-benzamide

I l l
1-16. n is 1 and R2o is piperidine
This intermediate is prepared from 1-2, via the procedure described above for 1-15. LCMS: m/z 376.12 (M+ 1).
I- 17. n is 1 and R2O is pyrrolidine This intermediate is prepared from 1-3, via the procedure described above for 1-15. LCMS: m/z 362.1 1 (M+I ).
1-18. n is 1 and R2o is morpholine
This intermediate is prepared from 1-4, via the procedure described above for 1-15. LCMS: TR = 1.35 min m/z 378.12 (M-H ). 1-19. n is 1 and R20 is N(CH3)2
This intermediate is prepared from 1-5, via the procedure described above for 1-15. LCMS: m/z 336.12 (M+ 1).
1-20. n is 1 and R20 is CH2N(CH3),
This intermediate is prepared from 1-6, via the procedure described above for 1-15, LCMS: m/z 350.10 (M+l).
1-21. π is 1 and R20 is CH2OCH3
This intermediate is prepared from 1-7, via the procedure described above for 1-15. LCMS: TR = 1.18 min m/z 337.08 (M+l).
EXAMPLE 3. N-ALKYLATED ANALOGS OF N-[I -(3,4-DIMETHYL-BENZYL)-PIPERIDIN- 4-YL]-3,5-DIMETHOXY-BENZAMϊDE
Intermediates prepared as described In Example 2 are reductively alkylated with any variety of agents to generate compounds of the formula:

69. n is 1 and R2o = pyrrolidine
To a solution of intermediate 1-17 (14.4 mg, 40 μmol) and 3.4-dimethylbenzaldehyde (8 mg, 60 μmol) in toluene (1000 μL) and acetic acid (50 μL) is added NaBH(OAc)3 (21.2 mg, 100 μmol). The mixture is stirred at 50 0C overnight. The reaction mixture is cooled to rt and quenched with 500 μL of IN NaOH. The organic phase loaded onto SCX cartridge (0.5 g of resin) which is pre-
washed with MeOH (3 mL), and eluted with MeOH (3 mL) to remove non basic impurities, and EtOAc: MeOH: TEA (10: 2: 1 , 3 mL) to afford the title compound as a yellow oil. LCMS: TR = 2.31 min, m/z 480.37 (M+I).
70. n is 1 and R20 = pipeiϊdine This compound is prepared from intermediate 1-16 via the procedure described above.
LCMS: TR = 2.33 min, m/z 494.39 (M+l).
71. n is 1 and R2ø - morpholine
This compound is prepared from intermediate 1-18 via the procedure described above. LCMS: TR = 2.35 min, m/z 496.39 (M+l). 72. n is 0 and R20 = piperidine
This compound is prepared via the procedure described above. LCMS: TR = 2.35 min, m/z 480.37 (M+l).
73. n is i and R20 = methoxy
This compound is prepared via the procedure described above. LCMS: TR = 1.40 min, m/z 441.18 (M+l).
74. n is 1 and R20 = CH2OCH3
This compound is prepared from 1-21 via the procedure described above. LCMS: TR = 1.42 min, m/z 455.18 (M+I).
75-78.
 PHENYL)-ETHYL]-PIPERIDIN-4-YL} -BENZAMIDE
PHENYL)-ETHYL]-PIPERIDIN-4-YL} -BENZAMIDE
Intermediates prepared as described for above are reductively alkylated with any variety of agents to generate compounds of the formula:

The following representative compounds are prepared in this manner:
75. 3s5-Dimethoxy-N-{ l-[l-(4-methoxy-233-dimethyl-phenyl)-etliyl]-piperidin-4-yI}-N-(2- methoxy-ethyl)-benzamide

A mixture of 3,5-dimethoxy-N-(2-methoxy-ethy])-N-ρiperidin-4-yl-benzamide (64.4 mg, 0.2 mmol, ] eq.) and ] -(4-methoxy-2,3-dimethyl-phenyl)-ethanone (35.6 mg, 0.2 mmoi, 1 eq.) in Ti(OiPr)4 (1 17.2 μL, 0.4 mmol, 2 eq.) is heated under N2 at 70 0C for 3 h. The reaction mixture is cooled to 0 0C, and anhydrous EtOH (10 mL) is added, followed by NaBH4 (10.8 mg, 0.3 mmol. 1.5 eq.) portionwise. The mixture is stirred at rt overnight. The reaction is quenched by the addition of sat. Na2CO3 (25 mL), and extracted with Et2O (3 x 25 mL). The organic layers are combined, washed with IN NaOH (50 mL), water (50 mL) and brine (50 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to afford the title compound as a yellow oil. LCMS: TR = 1.31 min, m/z 485.79 (M+ 1 ).
76. R2O is pyrrolidine
This compound is prepared via the procedure described above. LCMS: TR = 1.08 min, m/z 524.34 (M+l).
77. R2O is piperidine
This compound is prepared via the procedure described above. LCMS: TR = 1.21 min, m/z
538.21 (M+l).
78. R20 is morphoϋne This compound is prepared via the procedure described above. LCMS: Ts = 1.22 min, m/z
540.22 (M+ 1 ).
79. N-(l-{ l-[4-(3-HYDROXY-PROPOXY)-2,3-DIMETHYL-PHENYL]-ETHYL}-PIPERJD^N-4-YL)-3,5- DIMETHOXY-N-(2-METHOXY-ETH YL)-BENZAM1DE

2,3-dimethyl-phenyl)-ethyl]-piperidin-4-yl}-3,5-dimethoxy-N-(2-methoxy-ethyl)-benzamide (43 mg, 91.5 μmol, 1 eq.) in DMF (2 mL). The reaction is allowed to stir at rt for 30 min. 3-
Bromopropoxy-/_?Λ'/-butyldimethylsilaπe (42.4 μL, 1.83 mmol, 2 eq.) is added and the reaction is allowed to heat at 50 0C for 16 h. The reaction is quenched with IN NaOH (2 mL) and extracted with EtOAc (3 x 5 rnL). The organic layers are combined, washed with IN NaOH (5 mL), water (5 mL) and brine (5 mL), dried over Na2SO^, filtered and concentrated under reduced pressure. The resulting brown oil is purified by PTLC (100% CH2CL) to afford the desired protected intermediate.
This intermediate is dissolved in THF (500 μL) and cooled to 0 0C while IM TBAF (84 μL.. 2 eq.) is slowly added to the stirring solution. The reaction is allowed to slowly warm to rt and stirring is continued for 4 h. The reaction is quenched with IN NaOH (2 mL) and extracted with EtOAc (3 x 5 mL). The organic layers are combined, washed with water (5 mL) and brine (5 mL), dried over Na2SO1J, filtered and concentrated under reduced pressure. The resulting brown oil is purified by
PTLC (95: 5 CH2CI2: MeOH) to afford the title compound. LCMS: TR = 1.34 min, m/z 529.15
(M+ 1).
80. N-tl -I I -^^-HYDROXY-PROPOXYH^-DIMETHYL-PHENYLl-ETHYLJ-PIPERlDIW^-YL)^^- DIMETH0XY-N-(3-METH0XY-PR0PYL)-BENZAMIDE

NaH (35.8 mg, 1.49 mmol, 2 eq.) is added as a solid to a solution of N-{ l -[l-(4-Hydroxy-
2,3-dimethyl-pheny])-ethyI]-ρiρeridin-4-yl}-3,5-dimelhoxy-N-(3-methoxy-propyl)-benzamide (36 mg, 74.4 μmol, 1 eq.) in DMF (2 mL). The reaction is allowed to stir at rt for 30 min. 3- BiOmopropoxy-teπf-butyldimethylstlane (34.5 μL, 1.49 mmol, 2eq.) is added and the reaction is allowed to heat at 50 0C for 16 h. The reaction is quenched with IN NaOH (2 mL) and extracted with EtOAc (3 x 5 mL). The organic layers are combined, washed with IN NaOH (5 mL), water (5 mL) and brine (5 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting brown oil is purified by PTLC (100% CH2Ci2) to afford the desired protected intermediate. This intermediate is dissolved in THF (500 μL) and cooled to 0 0C while 1 M TBAF (61 μL, 2 eq.) is slowly added to the stirring solution. The reaction is allowed to slowly warm to rt and stirring is continued for 4 h. The reaction is quenched with IN NaOH ( 2mL) and extracted with EtOAc (3 x mL). The organic layers are combined, washed with water (5 mL) and brine (5 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting brown oil is purified by PTLC (95: 5 CH2Cl2: MeOH) to afford the title compound. LCMS: TR = 1.36 min, m/z 543.17 (M+l).
81. 3 -C HLQRO-4 -FLU ORO -N- { 1 -[ 1 -(4 -METHOX Y-2 , 3 -DIMETHYL-PHENYL)-ETHYL] -PIPERIDIN-4 - YL}-N-(2-METHOXY-ETHYL)-BENZAMIDE
Step 1 . { l -[l -(4-Methoxy-2,3-dimethyl-phenyl)-ethyl]-piperidin-4-yl}-(2-methoxy-ethyl)-amine


{ 1 ~ [ 1 -(4-Methoxy-2 , 3 -d im c y ! -pheny I)-ethy 1] -piper idi n-4-y 1 } -(2 -methoxy -ethy I )-am ine (9.6 mg. 30 μmol, 1 eq.) is dissolved in 400 μL of toluene and added to a 10% K2CO3 (750 μL). 3- chloro-4-fiuorobenzoyl chloride (8.6 mg, 45 μmol, 1.5 eq) is added and the solution is vigorously stirred at rt for 8 h. The reaction mixture is diluted with 1.0 mL EtOAc and the organic layer is separated. The organic layer is purified by SCX chromatography (0,5 g) to provide the title compound. LCMS: TR =1.39 min, m/z 459.20 (M+l).
82. 3 -FLUORO-N-[9-(4-METHOXY-2.3-DIMETHYL-BENZYL)-3-OXA-9-AZA-BICYCLO[3.3.1 ]NON-7- YL]-BENZΛMIDE

83. 3-FLUORO-N-[9-(2-HYDROXY-4,6-DIMHTHOXY-BENZYL)-3-OXA-9-AZA-BICYCLO[3.3.1 ]NON-7- YL]-BENZAMIDE

Stock solutions of the following materials are prepared: 3-fluoro-N-(3-oxa-9-aza- bicyclo[3.3.1]non-7-yi)-benzamide (0.1M in DMA); acetic acid (0.2M in DMA); sodium triacetoxyborohydride (0.2M in toluene); and 2-hydroxy-4,6-dimethoxy-benzaldehyde (0.2M in toluene). 0.2 mL of the amine, 0.2 mL of the aldehyde and 0.2 mL of the acetic acid solutions are combined and incubated together at 80 0C for 30 min, and then treated with 0.4 mL of the boro hydride solution. This mixture is incubated at 80 0C for 4 h. and then allowed to stand at rt for 18 h. The mixture is concentrated in vacuo and the residue is partitioned between EtOAc and saturated NaHCO3 solution. The organic layer is applied directly to an SCX cartridge and washed successively with 3 mL EtOAc, 3 mL 10: 1 EtOAc:MeOH and 10: 1 : 1 EtOAc:MeOH:TEA. The basic extract is concentrated in vacuo to give the title compound. LCMS (Method 3) m/z (M+H): 431.20, TR = 2.27 min.
84. ^C-N-[9-(l-{2,3-DIMETHYL-4-[(5-OXOMORPHOLIN-2-YL)METHOXY]PHENYL}ETHYL)-3-OXA- 9-AZABICYCLO[3.3.1]NON-7-YL]-3-FLUOROBENZAMIDE


2-(2,2-diethoxy-ethoxy)-Ll -diethoxy-ethane (19.7 g, 78.7 mmol, 1.0 eq.) is dissolved in 33 mL of 0.12M HC! and stirred at rt overnight, rac l-(4-Methoxy-2,3-dimethy!-phenyl)-ethy!amine HCl (14.1 g, 78.7 mmol, 1.0 eq.), sodium acetate (2.9 g, 35.4 mmol, 0.45 eq.), water (70 mL) and 3- oxo-pentanedioic acid (9.65 g, 66.1 mmol, 0.84 eq.) are added and the mixture is stirred at rt for 20 h and at 50 0C for 1 h. The acidic solution is cooled in an ice bath and basified to pH 12 - 14 with I OM NaOH. This is then extracted with EtOAc (4 x 300 mL). The combined extracts are washed with brine, dried over Na2SO^ and concentrated in vacuo leaving a dark, viscous oil. This is subjected to silica gel column chromatography eluting with 50% hexanes/EtOAc to provide the title compound as a reddish-orange foam. 1H NMR (300 MHz, CDCl3): δ 7.34 (m, IH), 6.72 (d, J=8.74 Hz, I H), 4.21 (m, IH), 3.90-3.76 (m, 3H), 3.80 (s, 3H), 3.70-3.50 (m, 4H), 3.04 (d, J=5.07 Hz, IH), 2.72-2.50 (m, 2H), 2.33 (m, 4H), 2.20-2.10 (m, 5H). LCMS (Method 3) m/z (M+H): 304.1, TR = 2.80 min. Step 2. rαc-9-[l-(4-Methoxy-2,3-dimethylphenyl)ethyl]-3-oxa-9-azabicyclo[3.3.1]nonan-7-one oxime

9-[I -(4-Methoxy-2,3-dimethylphenyl)ethyl]-3-oxa-9-azabicyclo[3.3.1 ]nonan-7-one (8.0 g, 26.4 mmol) in 65 mL of EtOH is treated with hydroxylamine hydrochloride (2.2 g, 31.6 mmol, 1.2 eq.) and stirred at rt for 90 h. The mixture is concentrated in vacuo and the residue is partitioned between 300 mL EtOAc and 100 mL water. The phases are separated and the organic phase is washed with brine, dried over Na2SO4, decanted and concentrated in vacuo leaving the title compound as a white foam. 1H NMR (300 MHz, CDCl3): δ 7.63-7.19 (m, 2H), 6.73 (d, J= 8.55 Hz, I H), 4.23 (d, J=6.01 Hz, I H), 3.96-3.82 (m, 2H), 3.82-3.56 (m, 4H), 3.81 (s, 3H)3 3.31 (bs, IH), 3.06 (dd, J=16.0 Hz, J=28.5 Hz, IH), 2.81 (bs, IH), 2.73-2.52 (m, IH), 2.36-2.25 (m, 5H),. 2.17 (s, 3H)5 1.28 (d, J=5.0 Hz, 3H). LCMS (Method 3) m/z (M+H): 319.1, TR = 2.73 min.
Step 3. rac-9-[l-(4-Methoxy-2,3-dimethylphenyl)ethyl]-3-oxa-9-azabicycIo[3.3.1 ]nonan-7-amine
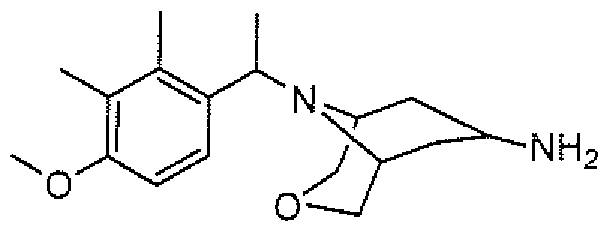
Step 4. rac-3-Fluoro-N-{9-[l-(4-methoxy-2,3-dimethylphenyl)-ethyl]-3-oxa-9- azabicyclo[3.3.1 ]non-7-yi} benzamide

Step 5. rac-3 -F luoro-N- { 9- [ 1 -(4-hydroxy-2,3 -d imethy lpheny l)-ethyl] -3 -oxa-9- azabicyclo[3.3. l]non-7-yl} benzamide


85. ΩC-N-(9- { L -[2,3 -DIMETHYL-4-(MORPHOLIN-2-YLMETHOXY)PHENYL]-ETHYL}-3-OXA-9- ΛZABICYCLOP .S .

3.0 eq.) and rac-fert-butyl 2-({[(4methylphenyi)sulfonyl]oxy}methyl)-morpholine-4-carboxylate
(150 mg, 0.41 mmol. 1.5 eq.) and stirred at rt for 46 h. The mixture is partitioned between EtOAc and water. The aqueous phase is extracted with EtOAc and the combined organic extracts are washed with water and brine, dried over Na2SO^ decanted and concentrated in vacuo. The residue is purified by preparative TLC (2 mm silica gel) eluting with 5% MeOH (2M NH3)/DCM to provide rac-tert-butyl 2-{ [4-( 1 -{7-[(3-fluorobenzoyl)aniino]-3-oxa-9-azabicyclo[3.3.1 ]nσn-9-yl}ethyl)-2,3 - dimethyi-phenoxy]methyl}morpholine-4-carboxylate as a foam. LCMS (Method 3) m/z (M+H):
612.27, TR = 2.89 min. This material is dissolved in 10 mL DCM, 5 mL MeOH, and 10 mL 4N HCI
in dioxane. After stirring for 3 h at it, the mixture is concentrated in vacuo and free based with saturated NaHCO3. After extracting with DCM, the solution is concentrated in vacuo and the residue is purified by preparative TLC using a 1 mm silica plate eluting with 10% MeOH (2M NH3)ZDCM to provide the title compound. !H NMR (300 MHz, CDC)3): δ 7.22-7. ϊ 3 (m, 3H), 6.74- 6.62 (td, J=LlO Hz, J=5.77 Hz5 J=2.47 Hz, I H), 6.08-5.99 (d, J=8.24 Hz1 IH), 5.16 (m, IH), 4.79 4.33 (bs, IH), 4.02-3.67 (m, 8H), 3.50-3.28 (m, SH)5 3.09 (bs, I H), 2.67 (bs, IH)3 2.33 (bs, 3H), 2.15 (s, 3H), 2.08-1.93 (m, 4H), 1.91-1.73 (m,3H), 1 .31 -1.1 1 (m, 3H). LCMS (Method 3) m/z (M+H): 512.18, TR = 2.22 min.
86. RAC-N-(8-{ 1 -[2,3-DlMETHYL-4-((2Δ}-5-OXOPYRROLIDIN-2-YLMETHOXY)-PHENYLJ-ETHYL}-8- AZA-BICYCLO[3.2.1]OCT-3-YL)-3-FLUOROBENZAMJDE


A solution of 2.5-dimethoxytetrahydrofuran (6.0 g, 45.6 mmol) in 12.5 mL of water is treated with concentrated HCl (2.2 mL, 26.4 mmol, 0.60 eq.). In a separate flask, a solution of rac- l-(4-methoxy-2,3-dimethyl-phenyl)-ethylamine (9.4 g, 57.0 mmol, 1.25 eq.) in 26 mL of water is cooled to 0 0C and treated with 6.7 mL concentrated HCl (80.4 mmol, 1.8 eq.) and stirred vigorously for about 20 min. The 2,5-dimethoxytetrahydrofuran solution previously prepared is diluted with an additional 18 mL water add is treated with the solution of l-(4-methoxy-2,3-dimethyl-phenyl)- ethylamine HCΪ. To this mixture is added a solution of 1.3 acetone dicarboxylic acid (7.33 g, 50.2 mmol, 1.1 eq.) in 29 mL and a solution of Na2HPC^ (3.23 g, 22.8 mmol, 0.5 eq.) in 15 mL water. The pH of the resulting solution is adjusted to -4.5 using 40% NaOH. This is then stirred at it overnight. The solution is then acidified to 7.5 with 6N HCl and heated in an 85 0C oil bath for 2 h. After cooling to rt, the solution is basified to pH 12 with 40% NaOH and extracted with DCM (3 x 200 mL). The combined organic extracts are washed with brine, dried over MgSO.^ filtered and concentrated in vacuo. The residue is purified by silica ge! column chromatography eluting with 1 :3:4% EtOAc :hexanes: TEA to provide the title compound. 1H NMR (300 MHz, CDCU): δ 7.18 (d, 1=9.14 Hz5 IH), 6.78 (d, J=9.2I Hz5 IH)5 3.82 (s, 3H), 3.75-3.62 (m, 2H), 3.48 (m, IH), 2.78-2.61 (m, 2H), 2.22 (s, 3H), 2.21-1.90 (m, 5H), 1.55 (m, 2H), 1.38 (d, J=5.71 Hz, 3H). LCMS (Basic method) m/z (M+H): 289.17, TR = 2.00 min. Step 3. rac- S-tl^-methoxy^^-dimethylphenyl^thylJ-S-azabicyclop^.ljoctan-S-one oxime

A solution of rac- 8-[l-(4-methoxy-2,3-dimethylphenyl)ethyi]-8-azabicyclo-[3.2.1]octan-3- one (12.0 g, 4 L7 mmol) in 100 mL EtOH is treated with hydroxylamiπe hydrochloride (3.5 g, 50.1 mmol, 1.2 eq.) and sodium bicarbonate (4.2 g, 50.1 mmol) and stirred at rt for 67 h. The mixture is concentrated to dryness, and the residue is suspended in 100 mL EtOAc and concentrated in vacuo. The residue is suspended in 200 mL diethyl ether and washed with 80 mL IN NaOH. The basic phase is extracted with EtOAc (2 x 100 mL). The combined organic extracts are washed with brine, dried over NaaSO^, decanted and concentrated in vacuo leaving the title compound as an amorphous white solid. 1H NMR (300 MHz, CDCl3): δ 8.19 (bs. I H)1 7.41 (bs, IH), 6.76 (d, J=8.52, IH). 3.93 (d, J=5.77, I H), 3.79 (s. 3H), 3.45 (bs, 2H), 2.88 (d, J=15.66 Hz, IH), 2.88 (td, J-2.47 Hz. J=IS- I l, IH) 2.34-2.25 (bs, 3H), 2.21-2.15 (bs, 4H), 2.09-1.84 (m, 4H), 1.75 (m, IH), 1.66-1.40 (m. 3H).
1.2S (d, J=6.04 Hz, 3H). LCMS (Method 3) mfz (M+H): 303.19, TR = 2.04 min. and 2.14 min. (syn and anti oximes).
Step 4. rac- 8-[l -(4-Methoxy-2,3-dimethylphenyl)ethyl]-8-azabicyclo[3.2.1]octan-3-one amine

A solution of rac- 8-[l-(4-methoxy-2,3-ciimethylphenyl)ethyl]-8-azabicycio[3.2.1]octan-3- one oxime (12.2 g, 40.3 mmol) in 200 mL /7-propanol is heated in a 70 0C oil bath and treated with metallic sodium (9.3 g, 403.4 mmol) added portionwise over 20-30 min. The mixture is heated to 120 0C (reflux) for 3 h. An additional 1 g of sodium is added and heating continued at reflux for another hour. The solution is cooled and the pH is cautiously adjusted to ~9 with concentrated HCl. The solvent is removed in vacuo and the residue partitioned between EtOAc and water. The organic extracts are dried and evaporated to give the title compound as a light yellow oil. 1H NMR (300 MHz, CDCl3): δ 7.50-7.33 (bs, I H), 6.72 (d, J=8.72 Hz, IH), 3.86 (s, IH), 3.79 (s, 3H), 3.40 (s, I H), 3.20 (s, IH), 2.28 (bs, 3H), 2.15 (s, 3H), 2.08-1.78 (m, 7H), ] .70-1.41(m, 8H), 1.29-1.19 (m, 5H). LCMS (Method 3) m/∑ (M+H): 289.32, TR = 1.78 min.
Step 5. rαc-3-Fluoro-N-{8-[l-(4-methoxy-2,3-dimethylphenyl)ethyl]-8-azabicyclo[3.2.1]oct-3- yljbenzamide

Rac-Z-{ I -(4-methoxy-2,3-dimethyiphenyl)ethyl]-8-azabicyclo[3.2. l ]octan-3-one amine (1.88 mg, 6.52 mmol), 3-fiuorobenzoyl chloride (940 μL, 7.82 mmol, 1.1 eq.) and potassium carbonate (1.35 g, 9.78 mmol, 1.5 eq.) are stirred together at rt in 60 mL Me-THF for 24 h. The solution is washed with water and brine, dried over Na2SO4, decanted and concentrated in vacuo. The residue is purified by silica gel column chromatography eluting with 5-10% MeOH (2M MH3)/DCM to provide the title compound as a white foam. 1H NMR (300 MHz, CDCl3): δ 7.63- 7.30 (m. 4H), 7.17 (m. IH), 6.73 (d, J=8.52 Hz, IH), 6.01 (bs, IH), 4.35 (bs, IH), 3.81 (s, 4H). 3.59 (bs, 3H), 3.23 (bs, IH), 2.41 -2.21 (bs, 3H), 2.17 (s, 3H), 2.14-2.00 (m, IH), 2.00-1.85 (m, 2H), 1.85- 1 .54 (m,5H), 1.28 (d, J=6.04 Hz, 3H). LCMS (Method 3) m/z (M+H): 41 1.17, TR = 2.47 min.
Step 6. Preparation of /m>3-fluoro-N-{8-[l -(4-hydroxy-2,3-dimethyl-phenyI)ethyl]-8- azabicycio[3.2.1]oct-3-yl}benzamide

[3.2.1]oct-3-yI}benzamide hydrochloride (prepared by treating 2.34 g, 5.70 mmol of the free base with 3 eq. IM HCl in diethyl ether followed by concentration in vacuo) in 40 mL of anhydrous DCM and cooled to <-70 0C under a nitrogen atmosphere. This solution is treated with 23.0 mL (23.0 mmol, 4.0 eq.) of IM BBr3 in DCM, which is added dropwise via syringe over 20 min. The mixture is allowed to warm to rt over 66 h and then diluted with 400 mL CHCl3 and 300 mL saturated NaHCO3 and vigorously stirred for 60 min. The phases are separated and the aqueous phase extracted with 300 mL CHCl3. The combined organic extracts are dried over Na2SO4, decanted and concentrated in vacuo to give the title compound. 1H NMR (300 MHz, CDCl3): δ 7.60-7.33 (m, 3H) 7.19 (m, IH), 6.66 (d, J=8.52 Hz, IH), 5.98 (d, J=7.97 Hz, IH), 4.34 (m, I H), 3.77 (bs, IH), 3.52 (bs, IH), 3.18 (m, IH), 2.27 (s, 3H), 2.18 (s, 3H), 2.20-1.49 (m, 8H), 1.24 (d, J=6.04 Hz, 3H). LCMS (Method 3) m/∑ (M+H): 397.06, Ta = 2.20 min.
Step 7. rαc-N-(8-{ l-[2,3-Dimethyl-4-((25)-5-oxopyrrolidin-2-ylmethoxy)-phenyl]-ethyl}-8-aza- bicyclo[3.2.1]oct-3-yl)-3-fluorobenzamide

A solution of rαc-3-chloro-4-fluoro-N-{8~[l-(4-hydroxy-2,3-dimethyl-phenyl)ethyl]-8- azabicyclo[3.2.1]oct-3-yI}benzamide (160 mg, 0.40 mmol), [(2i?)-5-oxopyrroIidin-2-y]]methyl A- methylbenzenesulfonate (135 mg, 0.50 mmol, 1.25 eq.), cesium carbonate (195 mg, 0.60 mmol, 1.5 eq.) and tetra-butyl ammonium iodide (15 mg, 0.04 mmol, 10%) in 3 mL DMF are stirred together at rt for 68 h. The product is precipitated by the addition of 12 mL water and collected by filtration to provide the title compound. 1H NMR (300 MHz, CDCi3): δ 7.54-7.31 (m, 3H), 7.18 (m, IH), 6.66 (d, J=8.24 Hz, IH), 5.90 (bs, I H), 4.43-4.25 (m, I H) 4.10 (m, IH), 3.97 (dd, J=3.30 Hz, J=9.34 Hz, IH), 3.88-3.72 (id, J=1.37 Hz, J=9.34 Hz, IH), 3.51 (bs, IH), 3.18 (bs, IH), 2.52-2.18 (m, 6H), 2.15 (s, 3H), 2.10-1.85 (m, 4H), 1.85-1.42 (m, 8H), 1.24 (d, J=6.32 Hz, 3H). LCMS (Method 3) m/z (M+H): 494.30, TR = 2.23 min.
87. AJC-N-(S- { 1 -[23-DlMETHYL-4-((25)-5-OXOPYRROLIDIN-2-YLMETHOXY)-PHENYL]-ETHYL} - AZA-B1CYCLO[3.2.1 ]OCT-3-YL)-3-CHLORO-4-FLUOROBENZAMiDE


"uc-8-[! -(4-Methoxy-2,3-dimethylphciiyl)cthy]]-8-azabicyc]o[3.2.1]octan-3-orjC amine (880 mg, 3.] mmol), 3-chloro-4-fluorobenzoyl chloride (883 mg, 4.5 mmol, 1.5 eq.) and potassium carbonate (631 mg, 4.5 mmol, 1.5 eq.) are stirred together at rt in 10 mL DCM for 20 h. The solution is partitioned between EtOAc and water and the organic phase washed with brine. This is dried over Na2SO.j, decanted and concentrated in vacuo. The residue is purified by silica gel column chromatography eluting with 3.5%-5% MeOH (2M MH3)ZDCM to provide the title compound as an off-white foam. 1H NMR (300 MHz, CDCI3): δ 7.81 (dd, J-2.20, J=4.67, IH), 7.62 (m, IH), 7.42 (bs, IH), 7.18 (m, 2H), 6.72 (d, J=8.79 Hz, I H), 5.85 (bs, I H), 4.33 (m, I H), 3.82 S9 4H), 3.55 (bs, IH), 3.23 (bs, IH), 2.38-2.22 (bs, 3H), 2.17 (s, 3H), 2.1 1-1.85 (m, 4H), 1.84-1.51 (m, 6H), 1.25 (d, J=5.77 Hz, 3H). LCMS (Method 3) m/z (M+H): 445.21 , TR = 2.62 min.
Step 2. rac- 3-Chloro-4-fluoro-N-{8-[l-(4-hydroxy-2,3-dimethyl-phenyl)ethyl]-8- azabicycIo[3.2.1]oct-3-yl}benzamide

A solution of rαc-3-chloiO-4-fluoro-N-{8-[l-(4-methoxy-2,3-dimethylphenyl)ethyt]-8- azabicycio[3.2.1]oct-3-yi}benzamide hydrochloride (prepared by treating 1.23 g, 2.76 mmol of the free base with 3 eq. IM HCl in diethyl ether followed by concentration in vacuo) in 25 mL of anhydrous DCM and cooled to <-70 0C under a nitrogen atmosphere. This solution is treated with 12.0 mL (12.0 mmol, 4.0 eq.) of IM BBr^ in DCM, which is added dropwise via syringe over 10 min. The mixture is allowed to warm to rt overnight and then diluted with 50 mL CHCI3 and 50 mL saturated NaHCO3 and vigorously stirred for 60 min. The phases are separated and the organic phase is dried over Na^SO^ decanted and concentrated in vacuo to give the title compound. LCMS (Method 3) m/z (M+H): 431.08, TR = 2.40 min.
Step 3. røc-N-(8-{ l-[253-Dimethyi-4-((25)-5-oxopyrrolidin-2-ylmethoxy)-phenyl]-ethyl}-8-aza- b i cyclo[3.2.1] oct-3 -y l)-3 -ch ϊ oro-4-fluorobenzamide

88. A4C-N-{8-[I -(2,3-DIMETHYL-4-{[(2J?)-5-OXOPYRROLIDJN-2 -YL]METHOXY) -PHENYL)-ETHYL]- 8-ΛZΛBIC YCLO[3.2.1] OCT-3 -YL } -3 -FLUOROBENZAMIDE

This compound is prepared essentially as described above, using readily apparent modification of starting material, and is obtained as a white form. 1H NMR (300 MHz, CDCI3): δ 7.53-7.33 (m, 3H), 7.17 (m, IH), 6.67 (d, J=8.52 Hz, IH), 6.09 (d, J=7.42 Hz, IH), 6.04 (bs , IH), 4.35 (m, IH), 4.08 (m, IH), 3.97 (dd, J=3.85 Hz, J=9.34 Hz, 1H),3.82 (dd, J=I .99 Hz, J=7.69 Hz I H), 3.53 (bs, IH), 3.20 (bs, I H), 2.47-2.22 (m, 6H), 2.14 (s, 3H), 2.09-1.84 (m, 6H), 1 .84-1.52 (m, 5H). 1.24 (d, J=6.04 Hz, 3H). LCMS (Method 3) m/z (M+H): 494.34, TR = 2.21 min.
89. ΛΛC-N-{8-[l -(2,3-DIMETHYL-4-{[(2/.)-l -METHYL-5-OXO-PYRROLIDIN-2- YL]METHOXY)- PHENYLjETHYLJ-δ-AZABICYCLOβ .2. l ]OCT-3-YL)-3-FLUOROBENZAMIDE

1.24 (d, 1=6.04 Hz, 3H); LCMS (Basic method) m/z (M+H): 508.23, TR: 2.20 min.
90. ωC-N-{8-[l -(2,3-DIMETHYL-4-{[(25>l -METHYL-5-OXO-PYRROLIDIN-2-
YL] METHOX Y} PHEN YL)-ETH YL] -8- AZ ABICYCLO [3.2.1 ]OCT-3 -YL } -3 -FLUOROBENZ AMIDE

This compound is prepared essentially as described above, using readily apparent modification of starting material. 1H NMR (300 MHz, CDCl3): δ 7.63-7.31 (m, 4H), 7.16 (ddd, J=0.82 Hz, J=2.47 Hz, J=8.24 Hz, I H)5 6.69 (d, J=8.52 Hz, IH), 6.07 (d, J=7.69 Hz, IH), 5.27 (s, 2H), 4.35 (m,lH), 4.13-3.86 (m, 3H), 3.79 (bs, IH), 3.57 (bs, IH), 3.21 (bs, I H), 2.89 (s, 3H), 2.58 (m, I H), 2.49-2.17 (m, 6H), 2.14 (s, 3H), 2.10-1.83 (m, 5H), 1.83-1.51 (m, 6H), 1.26 (d, J=5.77 Hz, 3H); LCMS (Method 3) m/z (M+H): 508.26, TR - 2.23 min.
91. ΛΛC-4-CYANO-3 -FLUORO-N- { 8-[ 1 -(4-METHOXY-2,3-DIMETHYLPHENYL)ETHYL]-8- AZABICYCLOP ^J ]OCT-S -YL)BENZAMIDE


93. /WC-N-(4-CHLORO-3-METHOXYPHENYL)-8-[l-(4-METH-OXY-2,3 -DIMETHYLPHENYL)-ET1 IYL]-S- AZABIC YCLO[3.2.1 ]OCTAN-3 -AMINE

Step 1. 6-(hydroxymethyl)piperidin-2-one

The following is a general procedure for the reduction of esters to primary alcohols. Ethyl ό-oxopiperidine-2-carboxylate (2.5 g, 14.6 mrnol) is dissolved in a sofution of 2M
LiBH^THF (7.5 mL, 15.0 mmol) and the resulting mixture is stirred at rt for 18 h, during which it becomes a slurry. The reaction mixture is carefully quenched with MeOH to dissolve the solids and stirred for 20 min. The crude mixture is concentrated under vacuum, and then purified by flash silica gel chromatography (MeOHVCH3CI2 = 15:85) to afford the title compound as a clear oil. Step 2. (6-oxopiperidin-2-yl)methyl 4-methylbenzenesulfonate

The following illustrates a general procedure for the formation of alkyl tosylates.
To a solution of 6-(hydiOxymethyl)piperidin-2-one (1 .2 g, 9.3 mmo!) in 50 mL dichbromethane are added TEA (1 1.7 ml, 83.7 mmol) and DMAP (1 15 mg, 0.93 mmol). To the resulting solution is added /Moluenesulfonyl chloride (1.95 g, 10.2 mmol) in small portions. The resulting solution is stirred at rt for 18 h. The reaction mixture is then washed with H2O, saturated aqueous NaHCθ3, IN HCl, and brine, dried over MgSθ4, filtered, and concentrated under vacuum. Crystallization of the residue with 1 : 1 Hex:EtOAc affords the title compound as a yellow solid. LCMS (Method 3) m/z (M+H): 284.00. TR = 2.22 min.
1-23. (5-OXOPYRROLIDIN-3-YL)METHYL 4-METHYLBENZENESULFONATE

Step 1. l-benzyl-4-(hydroxyrnethyl)pyrrolidin-2-one

Step 2. 4-(hydroxymethyl)pyrrolidin-2-one

To a solution of l-benzyl-4-(hydroxymethy])pyrrolidin-2-one (1.9 g, 9.25 mmol) in 40 IΏL liquid NH3 at -5O0C is added sodium metal (2.15 g, 93.5 mmol) in small portions. The reaction mixture turns dark blue upon addition of the sodium. The reaction mixture is warmed to reflux (-33 0C) and stirred for 1 h, after which it is cooled to -78 0C and quenched with excess EtOH. The reaction mixture is slowly warmed to rt and stirred for 18 h. The reaction mixture is then deposited directly onto silica under vacuum. Purification by flash silica gel chromatography (MeOH/CH2Cl2 = 1 :4) affords the title compound as a light purple oil.
Step 3. (5-oxopyrroIidin-3-yl)methyl 4-methylbenzenesulfonate

1-24. 2-(2-OXOIMIDAZOLIDIN-l -YL)ETHYL 4-METHYLBENZENESULFONATE

A 75% aqueous solution of l-(2-hydroxyethyl)imidazolidin-2-one (15 g solution = 11.25 g starting material, 86.4 mmol) is suspended in 200 mL benzene and refiuxed under a Dean-Stark trap for 3 h to remove water. Additional benzene is added to the reaction flask as necessary. The solution is cooled to rt and concentrated under vacuum to give dehydrated starting material. The title compound is then prepared using the general procedure described above for the formation of alkyl tosylates, except for the purification method. The crude product is purified by flash silica gel chromatography (MeOHZCH2Cl2 = 1 :9) to afford the title compound as white solid. LCMS (Method 3) m/z (M+H): 284.98, TR = 2.10 min.
1-25. 2-(2-OXO-l,3-OXAZOLIDIN-3-YL)ETHYL 4-METHYLBENZENESULFONATE

The title compound is prepared from 3-(2-hydroxyethyl)-l,3-oxazolidin-2-one using the general procedure described above for the formation of alkyl tosylates, except for the purification method. The crude product is purified by flash silica gel chromatography (MeOHZCH2Ci2 = 5:95) to afford the title compound as white solid. LCMS (Method 3) m/z (M+H): 285.99, TR = 2.10 min.
1-26. 2-[2-(DlMETHYLAMINO)-5-METHYL- 1 J-THIAZOL-4-YLJETHYL 4- METHYLBENZENESULFONATE

Step I . Methyl [2-(dimethylamino)-5-methy!-l,3-thiazol-4-yl]acetate
 l,l-Dimethylthiourea (521 mg, 5.0 mmol) and methyl 4-bromo-3-oxopentanoate (840 mg, 4.0 mmol) are combined in 20 mL toluene and heated at reflux overnight. The reaction mixture is washed with H2O, brine, dried over MgSO4, filtered, and concentrated under vacuum. The crude product is purified by flash silica gel chromatography (Hex/EtOAc = 1 : 1) to afford the title compound as a yellow oil LCMS (Method 3) m/z (M+H): 235. ] 3, TR = 0.83 min.
l,l-Dimethylthiourea (521 mg, 5.0 mmol) and methyl 4-bromo-3-oxopentanoate (840 mg, 4.0 mmol) are combined in 20 mL toluene and heated at reflux overnight. The reaction mixture is washed with H2O, brine, dried over MgSO4, filtered, and concentrated under vacuum. The crude product is purified by flash silica gel chromatography (Hex/EtOAc = 1 : 1) to afford the title compound as a yellow oil LCMS (Method 3) m/z (M+H): 235. ] 3, TR = 0.83 min.
Step 2. 2-[2-(dimethylamino)-5-methyl-l,3-thiazol-4-yl]ethanol

The title compound is prepared from methyl [2-(dirnethyIamino)-5-rnethyI-l,3-thiazol-4- yljacetate using the general procedure described above for the reduction of esters to primary alcohols, except for the purification method. The crude reaction mixture is carefully quenched with H2O, made slightly basic (pH=8) by the addition of IN HCl, and then extracted several times with EtOAc. The combined organic extracts are dried over MgSO4, filtered, and concentrated under vacuum to afford the title compound as a yellow oil. LCMS (Method 3) m/z (M+H): 187.18, TR = 0.48 min.
Step 3. 2-[2-(dimethylamino)-5-metliyl-l,3-thiazoI-4-yi]ethyI 4-methylbenzenesulfonate

The title compound is prepared from 2-[2-(dimethylamino)-5-methyl-I,3-thiazol-4- yljethanol using the general procedure described above for the formation of alkyl tosylates, except for the purification method. The crude product is purified by flash silica gel chromatography (Hex/EtOAc = 1: 1) to afford the title compound as a yellow oil. LCMS (Method 3) m/∑ (M+H): 341.09, TR = 2.12 min.
1-27. 2-(2 , 5 -DIMETHYL- 1 ,3 -THI AZOL-4- YL)ETHYL 4-METHYLBENZENES ULFON ATE
 The title compound is prepared from 2-(2,5-dimethyl-ϊ,3-thiazol-4-yl)ethanol using the genera] procedure described above for the formation of alkyl tosylates, except for the purification method. The crude product is purified by flash silica gei chromatography (Hex/EtOAc = 1 : 1) to afford the title compound as a yellow oil.
The title compound is prepared from 2-(2,5-dimethyl-ϊ,3-thiazol-4-yl)ethanol using the genera] procedure described above for the formation of alkyl tosylates, except for the purification method. The crude product is purified by flash silica gei chromatography (Hex/EtOAc = 1 : 1) to afford the title compound as a yellow oil.
ϊ-28. 2-( 1 -METHYL- l H-PYRAZOL-5-YL)ETHYL 4 -METIIYLBENZEM E SULFONATE

The title compound is prepared from 2-(l -methyl- lH-pyrazoI-5-yi)ethanol using the general procedure described above for the formation of alkyl tosylates, except for the purification method. The crude product is purified by flash silica get chromatography (Hex/EtOAc = 1:3) to afford the titie compound as a clear oil. LCMS (Method 3) m/z (M+H): 281.08, TR = 2.34 miπ.
1-29. 3-CHLORO-N-{8-[l-(2,3-DIMETHYL-4-{[(2.ft)-5-OXOPYRROLlD™-2-
YL]METS 1OX Y} PHEN YL)ETHYL]-8-AZABIC YCLO[3.2.1 ]OCY-3-YL}-4-FLUOROBENZAMIDE

The following illustrates a general method for the alkylation of a phenol with an alkyl tosylate or alkyl halide.
To a solution of 3-chloiO-4-fIuoro-N-{8-[l-(4-hydroxy-2,3-dimethylphenyl)ethyi]-8- azabicyclo[3.2. I]oct-3-yl}benzamide (65 mg, 0.15 mmoi) and [(2R)-5-oxopyrrolidin-2-yl]methyI 4- methylbenzenesulfonate (80 mg, 0.30 mmol) in 1 mL DMF is added CS2CO3 (145 mg, 0.45 mmol). The resulting mixture is stirred at it for 1 S h. The reaction mixture is diluted with 50 mL EtOAc and then washed with H2O, and then with brine, dried over MgSθ4, filtered, and concentrated under vacuum. The crude product is purified by PTLC (MeOH (2M NH3)/CH2C12 = 1 :9) to afford the title compound as a yellow oil. LCMS (Method 3) m/z (M+): 528.24, TR = 2.40 min.
94. 3-CHLORO-N- {8-[ 1 -(2,3-DIMETHYL-4-{[(25)-5-OXOPYRROLIDIN-2- YLJM£THOXY}PHENYL)ETHYL]-8-AZABICYCLO[3.2.1]OCT-3-YL}-4-FLUOROBENZAivIIDE

The title compound is prepared from the phenol and [(25)-5-oxopyrrolidin-2-yl]methyl 4- methylbertzenesulfonate using the alkylation procedure described above. LCMS (Method 3) m/z (M+): 528.25, TR = 2.39 min.
95. 3-CHLORO-N-[(/Λ,5iSr)-8-(2,3-DIMETHYL-4-{[(2J?)-5-OXOPYRROLIDIN-2- YL]METHOX Y} BENZYL)-8-AZABICYCLO[3.2.1 ]OCT-3-YL]-4-FLUOROBENZAMIDE

The title compound is prepared from the phenol and [(2ff)-5-oxopyrrolidin-2-yl]methyI A- methyibenzenesulfoπate using the alkylation procedure described above. LCMS (Method 3) m/z (M+): 514.23 , TR = 2.32 min.
96. 3-CHLORO-N-[(7J?,55)-8-(2,3-D!MEΪHYL-4-{[(2iS)-5-OXOPYRROL!DIN-2-
YL]METHOXY}BENZYL)-8-AZABICYCLO[3.2.1]OCT-3-YL]-4-FLUOROBENZAMlDE

The title compound is prepared from the phenol and [(25)-5-oxopyriOfidin-2-yl]methyl A- methylbenzenesulfonate using the alkylation procedure described above. LCMS (Method 3) m/z (M+): 514.34, TR = 2.32 min.
97. 3-CHLORO-N-[8-(l -{2,3-DIMETHYL-4-[(6-OXOPYRROLIDlN-2-YL)METHOXY]PHENYL}ETHYL)- 8-AZABICYCLO[3.2.1 ]OCT-3 -YL] -4-FLUOROBENZ AMIDE

The title compound is prepared from the phenol and (6-oxopiperidin-2-yl)methyJ 4- methylbenzenesulfonate (synthesis previously described) using the aikylation procedure described above, m/z 542.18 (M+). LCMS (Method 3) m/z (M+): 542.18, TR = 2.52 min.
98. 3-CHLORO-N-[8-(1 -{2,3-DIMETHYL-4-[(5-OXOPYRROLIDIN-3 -YL)METHOXY]PHENYL}ETHYL)-8- AZABICYCLO[3.2.1 ]OCT-3 -YL]-4-FLUOROBENZAMIDE

99. 3-CHL0R0-N-[8-( 1 -{2,3-DIMETHYL-4-[2-(2-OXOPYRROLIDIN- 1 -YL)ETHOXY]PHENYL) ETHYL)- 8-AZABICYCLO[3.2.1 ]OCT-3-YL]-4-FLUOROBENZAMEDE

The title compound is prepared from the phenol and 3-(2-chloroethyf)-1.3-oxazo!idin -2-one using the aikylation procedure described above. LCMS (Method 3) m/z (M+H): 508.31, TR = 2.30 min.
100. 3-CHLORO-N-[8-(l -{2.3-DIMErHYL-4-[2-(2-OXO-1.3-OXOZOLIDIN-3-
YL)ETHOXY]PHENYL}ETHYL)-8-AZABICYCLO[3.2.1]OCT-3-YL]-4-FLUOROBENZAMIDE

101. 3-CHLORO-N-[δ-(l-{2,3-DIMETHYL-4-[2-(2-OXOIMIDAZOLIDIN-l- YL)ETHOXY]PHENYL}ETHYL)-8-AZABICYCLO[3.2.1]OCT-3-YL]-4-FLUOROBENZAMIDE

The title compound is prepared from the phenol and 2-(2-oxoimidazoiidin-l-yl)ethyi 4 -methyi benzene sulfonate (synthesis previousϊy described) using the alkylation procedure described above. LCMS (Method 3) m/z (M+): 543.18, TR = 2.39 min.
102. 3 -CHLORO-N-[8-(l-{2,3-DIMETHYL-4-[3 -(2-OXOIMIDAZOLIDIN- 1-
YL)PROPOXY]PHENYL}ETHYL)-8-AZABICYCLO[3.2.1 ]OCT-3-YL]-4-FLUOROBENZAMIDE

The title compound is prepared from the phenol and ] -(3-chloropropyl)imidazoIidin-2- one using the aikyiation procedure described above. LCMS (Method 3) m/z (M+): 557.34, TR = 2.50 min.
103. N-(8-{ I -[4-(3-ACETAMIDOPROPOXY)^J-DIMETIIYLPHENYL]ETHYL) -8- AZABICYCLO[3.2.1]OCT-3-YL)-3-CHLORO-4-FLUOROBENZAMIDE


The title compound is prepared from the phenol and tert-butyl (3-bromopropyl)- carbamate using the alkylation procedure described above. LCMS (Method 3) m/∑ (M+): 588.50, TR = 2.76 min.
Step 2. N-(8-{ l-[4-(3-aminopropoxy)-2,3-dimethy]phenyi3ethyI}-8-azabicycIo[3.2.1]oct-3-yl)-3- chloro-4-fiuorobenzamide

/er/-Butyl-{3-[4-(l -{3-[(3-chloro-4-fluorobenzoyl)amino]-8-azabicyclo[3.2.1]oct-8-yl}- ethyl)-2,3-dimethylphenoxy]propyi} carbamate (300 mg, 0.5 ramol) is dissolved in 10 mL 10% CF3COOH/CH2CI2 and the resulting mixture is stirred at room temperature for 18 h. The reaction mixture is concentrated under vacuum, diluted with saturated aqueous NaIICO3 and then extracted several times with EtOAc. The combined organic extracts are washed with brine, dried over MgSθ4, filtered and concentrated under vacuum to afford the title compound as a clear oil. LCMS (Method 3) m/∑ (M+); 488.29, TR = 2.16 min.
Step 3. N-(8-{ l-[4-(3-acetamidopropoxy)-2.3-dimethylphenyl]ethyl}-8-azabicyclo[3.2.1]oct-3-y[)- 3-chloro-4-fiuorobenzamide
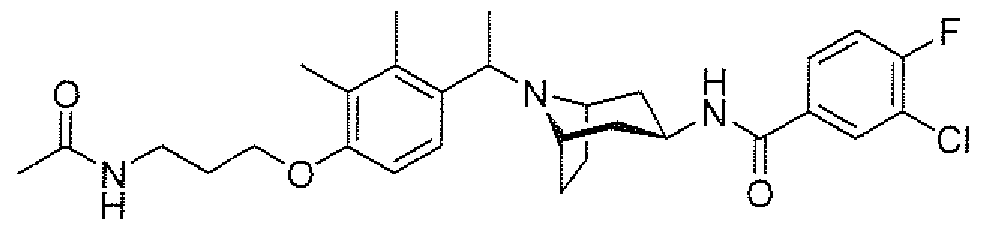
To a stirred solution of N-(8-{ l-[4-(3-aminoρropoxy)-2,3-dimethyiphenyi]ethyl}-8- azabicyclo[3.2.1]oct-3-yl)-3-chloro-4-fiuorobenzamide (50 mg, 0.1 mmol) and TEA (60 μL, 0.4 mmol) in 1 mL CH2CI2 is added acetyl chloride (22 μL, 0.3 mmol). After 1 h, LCMS shows a 1 : 1 ratio between the desired mono N-acylated product and the bis N-acy!ated product. The reaction mixture is concentrated under vacuum, diluted with 2 mL MeOH, IN NaOH is added (0.5 mL, 0.5 mmol), and the resulting mixture is stirred at rt for 18 h. The reaction mixture is diluted with H2O and extracted several times with EtOAc. The combined organic extracts are washed with brine, dried over MgSθ4, filtered and concentrated under vacuum. The crude product is purified by PTLC
(MeOH (2M NH3)/CH2C12 = 1 :9) to afford the title compound as a yellow oil. LCMS (Method 3) m/z (M+): 530.27; TR = 2.46 min.
104. REL-3 -CHLORO-N- {(1R, 3S)-&-[ 1 -(4- {2- [2-(DIMETΗYLAMINO>5 -METH YL- 1 ,3 -THIAZOL-4- YL]ETHOXY } -2,3 -DIMETHYLPHENYL)ETHYL]-S-AZABICYCLOP .2.1 ]OCT-3 -YL } -4- FLUOROBENZAMIDE

The title compound is prepared from the phenol and 2-[2-(dimethylamino)-5-methyl-l ,3- thiazol-4-yl]ethyl 4-methyIbenzenesulfonate (synthesis previously described) using the alkylation procedure described above. LCMS (Method 3) m/z (M+): 599.42, TR = 2.88 min.
105. 3-CHLORO-N-{ 8-[1 -(4-{2-[2-(DΪMETHYLAMΓNO)-5-METHYL- 1 ,3-THIAZOL-4-YL]ETHOXY} -2,3- DIMETHYLPHENYL)ETHYL]-8-AZABICYCLO[3.2.1]OCT-3-YL}-4-FLUOROBENZAMIDE

The title compound is prepared from the phenoi and 2-(2,5-dimethyl-] ,3-thiazol-4-yl)ethyl 4-methylbenzenesulfonate (synthesis previously described) using the alkylation procedure described above. LCMS (Method 3) m/z (M+): 570.38, TR = 2.85 min.
106. 3-CHLORO-N-[8-(l -(2,3-DIMETI
 YL)ETHOXY]PHENYL}ETHYL)-8-AZABICYCLO[3.2.1]OCT-3-YL]-4-FLUOROBENZAMIDE
YL)ETHOXY]PHENYL}ETHYL)-8-AZABICYCLO[3.2.1]OCT-3-YL]-4-FLUOROBENZAMIDE

107. 3-CHLORO-N-[8-( 1 - (4-[2-(3,5-DIMETHYL-1H-PYRAZOL-4-YL)ETHOXY]-2,3-
DIMETHYLPHENYL}ETHYL)-8-AZABICYCLO[3.2.1]OCT-3-YL]-4-FLUOROBENZAMIDE

108. 3 -CHLORO-N- { 8- [ I -(4- { [(2S)-I-H YDROX Y-3 -( 1 H-PYRAZOL- 1 -YL)PROP YL]OX Y} -2,3 - DMETHYLPHENYL)ETHYL]-8-AZABICYCLO[3.2.1 ]OCT-3-YL} -4-(IH-PYRAZOL- 1 -YL)BENZAMIDE
Step 1. 3-chloro-N-[8-(l -{2,3-dimethyl-4-[(2S)-oxiran-2-ylmethoxy]phenyl}ethyl)-8- azabicyclo[3.2.1 ]oct-3-yl]-4-fJuorobenzamide

The title compound is prepared from the phenol and (2iS3-oxiran-2-ylmethyl 4- methylbenzenesuifonate using the alkylation procedure described above. LCMS (Method 3) m/z (M+): 487.28, TR = 2.51 min.
Step 2. 3-chloro-N-{8-[1 -(4-{[(2S>2-hydroxy-3-(lH-pyrazol-1 -y])propyl]oxy}-2,3- dimethyIρhenyl)ethyI]-8-azabicyclo[3.2.1 ]oct-3-yl}-4-( I H-pyrazol- 1 -yl)benzamide

109. 3-CHLORO-N-(8- { 1 -[4-({(25)-3 -[ETΉYL(METHYL)AMINO]-2-HYDROXYPROPYL} OXY)-2,3-
DIMETHYLPHENYL]ETHYL) -S-AZABICYCLOP .2. l ]0CT-3-YL>4-FLUOROBENZAMΪDE

1 10. 3-CHLORO-4-FLUORO-N-{8-[Ϊ -(4-{3-[(2-HYDROXYETHYL)-(METHYL)AMINO]PROPOXY} -2,3- DIMETHYLPHENYL)ETHYL]-S-AZABFCYCLOP ^. l]0CT-3 -YL)BENZAMIDE
Step 1. 3-chloro-N-(8-{ l-[4-(3-chloroproρoxy)-2.3-dimethylphenyl]ethyl}-8-azabicyclo[3.2.1 ]oct- 3-yj)-4-fluorobenzamide

The title compound is prepared from the phenol and l-chloro-3-iodopropane using the alkylation procedure described above. LCMS (Method 3) m/z (M+): 507.313 TR = 2.76 min.
Step 2. 3-chloro-4-fluoro-N-{8-[l-(4-{3-[(2-hydiOxyethyl)(methyI)amino]propoxy}-253- dimethylphenylJethyij-S-azabicyclop^.IJoct^-yllbenzamide

To a stirred solution of 3-chloro-N-(8-{ l -[4-(3-chloropropoxy)-2,3-dimethyl- phenyl]ethyl}-8-azabicyclo[3.2.1]oct-3-yϊ)-4-fluorobenzamide (170 mg, 0.33 mmol) and 2- (methylamino)ethanol (81 μL, 1.0 mmol) in 3 niL DMA is added K2CO3 (139 mg, 1.0 mmol). The resulting mixture is heated at 80 0C in a sealed, screw-capped vial for 3 h. The reaction mixture is cooied to rt, diluted with J-^O, and extracted several times with EtOAc. The combined organic extracts are washed with brine, dried over MgSθ4, filtered, and concentrated under vacuum. The crude product is purified by PTLC (MeOH (2M NH3)ZCH2Cl2 = 1 :9) to afford the titie compound as a yeϊlow oil. LCMS (Method 3) m/z (M+): 546.40, TR - 2.07 min.
1 1 1. 3-CHLORO-4-FLUORO-N- {8-[1-(4- {[(25)-2-HYDROXY-PROPYL]OXY}-2.3- DIMETHYLPIIENYL)ETHYL]-S-AZAB]CYCLOP ^1 I ]OCTO-YL) BENZAMIDE
Step 1. N-{8-[l-(4-{[(25)-2-{[/e^-butyl(dimethyi)silyl]oxy}propyl]oxy}-2,3- dimethylphenyI)ethyl]-8-azabicyclop.2.I]oct-3-yl}-3-chloro-4-fluorobenzamide

Step 2. 3-chloro-4-fluoiO-N-{8-[l-(4-{[(2-S)-2-hydroxypropyl]oxy}-2,3-dimethylphenyl)etliyl]-8- azabicyclo[3.2,l]oct-3-yl}benzamide

To a stirred solution of N-{8-[l-(4-{[(25)-2-{[fert- butyl(dimethyI)silyl]oxy}propyI]oxy}-2,3-dimethylphenyϊ)ethyl]-8-azabicyclo[3.2.1]oct-3-yI}-3- chloro-4-fluorobenzamide (60 mg, 0.1 mmol) dissolved in 1 mL THF is added 1.0 M TBAF in THF (0.15 mL, 0.15 mmol). The resulting mixture is stirred at rt for 18 h. The crude product is concentrated under vacuum and purified by HPLC to afford the title compound as a clear oil. LCMS (Method 3) m/z (M+): 489.31, TR = 2.07 min.
1 12-1 16. PREPARATION OF CERTAIN CYANOIMIDO COMPOUNDS

1 12. 3-CHLORO-N-(8-{ l-[4-(3-{[(7£)-N-CYANOETHANIMIDOYL]AMINO}PROPOXY>2,3- DIM£THYLPHENYL]ETHYL} -8-AZABlCYCLO[3.2. l ]OCT-3-YL)-4-FLUOROBENZAMIDE

1 13. METHYL N-{3-[4-(l -{3 -[(3 -CHLORO-^FLUOROBENZOYL)AMINO]-S- AZABICYCLO[3.2.1 ]OCT-S-YL}ETHYLj-2,3-DIMETHYLPHENOXY]PROPYL}-Nr- CYANOIM1DOTH1OCARBAMATE

To a solution ofN-(8-{ l-[4-(3-aminopropoxy)-2,3-dimethylphenyi]ethyl}-8- azabicyclo-[3.2. l]oct-3-yl)-3-ch!oro-4-flυorobenzamide (synthesis previously described; 80 mg,
0.16 mmol) in 4 mL EtOH is added dimethyl cyanodithioimidocarbonate (27 mg, 0.16 mmol). The resulting mixture is heated at 60 0C in a sealed, screw-capped vial for 18 h. The reaction mixture is cooled to room temperature, concentrated under vacuum, and purified by PTLC (2% TEA in 1 :9
MeOH/CH2Cl2) to afford the title compound as a clear oil. LCMS (Method 3) m/z (M+): 586.35, TR = 2.60 min.
1 14. METHYL N-p-^-O-p-^-CHLORO^-FLUOROBENZOYLJAMTNOJ-S-
AZABICYCLO[3.2.1 ]OCT-8-YL}ETHYL)-2,3-DIMETHYLPHENOXY]PROPYL}-N'-
CYANO.MIDOCARBAMATE

1 15. 3-CHLORO-N-[8-(l -{4-[3-(N"-CYANO-N'-METHYLCARBAMIMIDAMIDO)PROPOXY]-
2J4ΗMETHYLPHENYL}ETHYL)-8-AZABICYCLG[3.2.1]OCT-3-YL]-4-
FLUOROBENZAMIDE

3 16. 3-CHLORO-N-[8-(l-{4-[3-(N"-CYANO-N,N-
DIMETHYLCARB AMIMID AMIDO)PROPOXY]-2,3-DlMETHYLPHENYL}ETHYL)-8-
AZABICYCLO[3.2.1 ]OCT-3-YL]-4-FLUOROBENZAMIDE

The title compound is prepared using the procedure described above, except that 2M dimethyϊamine in MeOH (2 mL, 4.0 mmol) is used. LCMS (Method 3) m/z (M+): 583.42, TR = 2.51 min.
1 17. 3-CYANO-N-{8-[H2,3-DIMETHYL-4-{[(2R)-5-OXOPYRROLIDIN-2- YL]METHOXY) PHENYL)ETHYL]-S-AZABICYCLO[S^ I ]OCT-S-YL)BENZAMIDE
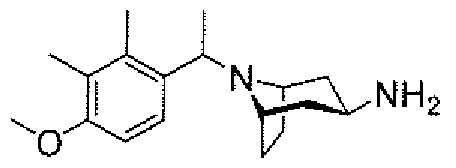 TEA, CH2CI2
TEA, CH2CI2


Step 1. 2,2,2-trifiuoro-N-{8-[l-(4-methoxy-2,3-dimethy]phenyl)ethyl]-8-azabicyclo[3.2.1]oct-3- yljacetamide

Step 2. 2,2,2-trifluoro-N-{8-[l -(4-hydroxy-2,3-dimethylpheny!)ethyI]-8-azabicyclo[3.2. l]oct-3- yl}acetamide

The following illustrates a general procedure for the demethylation of an aryl methyl ether using boron tribromide.
To a stirred solution of 2,2,2-trifluoro-N-{8-[I-(4-methoxy-2,3- dimethylphenyI)ethyl]-8-azabicyclo[3,2.1]oct-3-y]}acetamide (i l .3 g, 29.5 mmol) in 60 mL CH2CI2 is added 4N HCl in dioxane (1 1.1 mL, 44.4 mol). The resulting mixture is concentrated to dryness under vacuum. The resulting hydrochloride salt is re-dissolved in 100 mL CH2CL, cooled to -78 0C, and then treated with 3 M BBr3 in CH2Cl2 (89 mL, 89.0 mmol). The resulting mixture is warmed to rt and stirred for 3 h. The reaction mixture is carefully quenched with H2O, made basic (pH=8) by the addition of ION NaOH, and then extracted several limes with CH2Cl2. The combined organic extracts are washed with brine, dried over MgSθ4, filtered, and concentrated under vacuum. The crude product is purified by flash silica gel chromatography (MeOH (2M NH3VCH2Cl2 = 1 :9) to afford the title compound as a light yellow oil. LCMS (Method 3) m/∑ (M+H): 371.18, TR = 2.03 min.
Step 3. N-{8-[l -(2,3-dimethyl-4-{[(2i?)-5-oxopyrroiidin-2-yI]methoxy}ρhenyl)ethyl]-8- azabicyclo[3.2.1]oct-3-yl}-2,2,2-trifiuoroacetamide

The title compound is prepared from the phenol and Λ-(5-oxopyrrolidin-2-yi)methyl 4- methylbenzenesulfonate using the general alkylation procedure previously described. LCMS (Method 3) m/z (M+H): 468.28, TR = 2.09 min.
Step 4. (5Λ)-5-({4-[] -(3-amino-8-azabicyclo[3.2.1]oct-8-yl)ethyl]-2,3- dimethyIphenoxy}methyl)pyrrolidin-2-one

The following illustrates a general procedure for the alkaline hydrolysis of a trifluoroacetamide.
To a solution of N-{8-[I-(2,3-dimethyl-4-{[(.?iϊ)-5-oxopyrrolidin-2-yl]methoxy}- phenyI)ethy!]-8-azabicyclo[3.2.1]oct-3-yl}-2,2,2-trifiuoroacetamide (1.45 g, 3.1 mmol) in 15 mL MeOH is added ION NaOH (0.325 mL, 3.25 mmol). The resulting mixture is stirred at rt for 18 h, after which LCMS shows the presence of the desired product and unreacted starting material (approximately 3 : 1 ratio). An additional 0.325 mL I ON NaOH (3.25 mmol) is added, and the resulting mixture is heated at 60 0C for I S h. The reaction mixture is cooled to rt, concentrated under vacuum, diluted with IN HCl, and extracted with EtOAc (which is later discarded). The aqueous phase is made basic (pH=9-10) with I ON NaOH and then extracted several times with EtOAc. The combined organic extracts are washed with brine, dried over MgSO4, filtered, and concentrated under vacuum to give the title compound as a clear oil. LCMS (Method 3) m/z (M+H): 372.16, TR = 1.78 min.
Step 6. 3-cyano-N-{8-[] -(2,3-dimethyl-4-{[(2Jϊ)-5-oxopyrrolidin-2-yl]methoxy}phenyl)ethyi]-8- azabicyclo[3.2.1]oct-3-yl}benzamide

1 18. N-[8-(2,3-DIM£THYL-4-{[(2Λ)-5-OXOPYRROLIDlN-2-YL]METHOXY}BENZYL)-8- AZABTCYCLO[3.2.1]OCT-3-YL]-3-FLUOROBENZAMIDE

1 19. 3-CYANO-N-(8-{ l-[4-({(25)-3-[ETHYL(METHYL)AMlNO]-2-
HYDROXYPROPYL}OXY)-2,3-DIMETHYLPHENYL]ETHYL} -8-AZABICYCLOP ^. I]OCT- 3-YL)BENZAMIDE

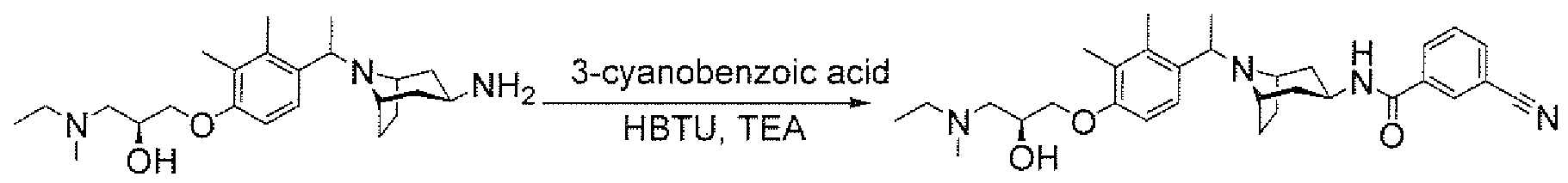
Step 1. N-[S-(I -{2,3-dimethyl-4-f(25)-oxiran-2-ylmethoxy]phenyl}ethyI)-8-azabicyclo[3.2.1]oct-3- yI]-2,2,2-trifluoroacetamide

The title compound is prepared from the phenol and (25)-oxiran-2~ylmethy! 4-methyl- benzenesulfonate using the alkylation procedure described above. LCMS (Method 3) m/z (M+): 487.31, TR = 2.48 min. Step 2. N-(8-{ l-[4-({(25)-3-[ethy](methy!)amino]-2-hydiOxypropyl}oxy>253- dimethylpheny]]ethyl}-8-azabicyclo[3.2.1]oct-3-yl)-2,2,2-trifluoroacetamide

To a solution of N-[S-(l-{2,3-dimethyl-4-[(2S)-oxiran-2-ylmethoxy]phenyI}ethyl)-8- azabicyclo[3.2.1]oct-3-yl]-2,2,2-trifJuoroacetamide (800 mg, 1.88 mmol) in 5 mL wo-propanol is added N-methylethanamine (0.8 mL, 9.4 mmol). The resulting mixture is heated at 80 0C in a sealed, screw-capped vial for 2 h. The reaction mixture is cooled to rt and concentrated under
vacuum to afford a yellow oil. The crude product is carried forward as is. LCMS (Method 3) m/z (M+H): 486.37, TR = 1.74 min.
Step 3. (25)-l-{4-[l-(3-amino-8-azabicyclo[3.2.1 ]oct-8-yl)ethy]]-2,3-dimethylpheπoxy}-3- [ethyl(methyl)amino]propan-2-ol

The title compound is prepared from N-(8-{ I-[4-({(25)-3-[ethyl(methyI)amino]-2- hydroxypiOpyl}oxy)-2,3-dimethylphenyl]ethyl}-8-azabicyclo[3.2.1]oct-3-yl)-2,2,2-trifluoro- acetamide using the general procedure described above for the alkaline hydrolysis of a trifluoroacetamide. The crude product is carried forward as is. LCMS (Method 3) m/z (M+H): 487.31, TR = 1.63 min.
Step 4. 3-cyano-N-(8-{ l-[4-({(2iS)-3-[ethyl(methyl)ammo]-2-hydroxypropyl}oxy)-2,3- dimethyiphenyI]ethyl}-8-azabicydo[3.2.] ]oct-3-y])benzamide

The title compound is prepared from (2S)- 1 -{4-[l -(3 -amino-8-azabicyclo[3.2. l]oct-8- yI)ethyl]-2,3-dimethylphenoxy}-3-[ethyl(methyl)amino]propan-2-ol and 3-cyanobenzoic acid using the previously described ΪIBTU coupling procedure. LCMS (Method 3) m/z (M +-H): 519.44, TR = 1.85 min.
120. 1 -(3-CHLOROPHEN YL)-3-[8-(4-METHOXY-2,3 -DIMETHYLBENZYL)-8- AZABICYCLO[3.2.1 ]OCT-3-YL]UREA

To a solution of 8-(4-sϊiethoxy-2,3-dimethylbenzyl)-8-azabicyc!o[3.2. ϊ]octan-3-amine (52 mg, 0.19 mmol) in 3 mL CH2Ci2 is added l-chloro-3-isocyanatobenzene (44 mg, 0.29 mmoi). The resulting mixture is stirred at rt for 30 min. The reaction mixture is then treated with excess Darco KB-BΦ powder and an amine scavenger resin and stirred for 1 h. The resin and Darco® powder are filtered off. and the solvent is removed under vacuum to afford the title compound as a yellow oil. LCMS (Method 3) m/z (M+H): 428.22, TR = 2.47 min.
121. 3-CHLORO-4-FLUORO-N-[8-(4-METHOXY-2,6-DJMETHYLBENZYL)-8- AZABICYCLO[S ^ I ]OCT-S-YL]BENZAMIDE
V A O r-


Step 1. fer/-Butyl [8-(4-methoxy-2,6-dimethylbenzyl)-8-azabicycio[3.2.1]oct-3-yI]carbaraate

The following illustrates a general procedure for the reductive amination of an amine with an aldehyde using sodium triacetoxyborohydride, and is referred to as Procedure 1 elsewhere herein.
To a solution of te?Y-butyl 8-azabicyclo[3.2.1]oct-3-ylcarbamate (400 mg, 1.77 mmol) and 4-methoxy-2,6-dimetiiylbenzafdehyde (406 rag, 2.47 mmol) in 10 ml CH2Cl2 is added sodium triacetoxyborohydride (524 mg, 2.47 mmol). The resulting mixture is stirred at rt for 18 h. The reaction mixture is washed with IN NaOH, brine, dried over MgSO4, filtered and concentrated under vacuum. The crude product is purified by flash silica gel chromatography (MeOHYCH2Cl2 = 1 :9) to afford the title compound as a clear oil. LCMS (Method 3) m/z (M+H): 375.38, TR = 1 ,37 min. Step 2. S-^-memoxy^ό-dimethylbenzyO-S-azabicycloP^. lJoctan-S-amine

The following illustrates a general procedure for the removal of a /-butyl carbamate group using hydrochloric acid.
To a solution of tert-buty] [8-(4-methoxy-2.6-dimethylbenzyl)-8-azabicyclo[3.2.1 ]oct-3- yl]carbamate (500 mg, 1.33 mmol) dissolved in 5 rriL CH2Cl2 is added 4N HCl in dioxane (4 mL,
16.0 mmol). The resulting solution is stirred at rt for 18 h. The reaction mixture is diluted with
EtOAc and extracted several times with IN HCl. The combined aqueous phases are collected, made basic (pH=9-10) with I ON NaOH. and extracted several times with Et2O. The combined ethereal extracts are dried over MgSO4. filtered, and the solvent is removed under vacuum to afford the title compound as a clear oil. LCMS (Method 3) m/z (M+H): 275.21, TR = 1.66 min.
Step 3. 3-chioro-4-f!uoro-N-[8-(4-methoxy-2,6-dimethylbenzyl)-8-azabicyclo[3.2.1]oci-3- yl]benzamide

The following illustrates a general procedure for the formation of an amide via the reaction of an amine with an acid chloride.
To a solution of 8-(4-methoxy-2,6-dimethylbenzyl)-8-azabicycIo[3.2.1]octan-3-amine (40 mg, 0.15 mmol) and TEA (40 μL, 0.29 mmol) in 1 mL CH2Cl2 is added 3-chloro-4-fiuorobenzoyl chloride (34 mg, 0.18 mmol). The resulting mixture is stirred at rt for 18 h. The reaction mixture is diluted with EtOAc, washed with saturated aqueous NaHCO3. brine, dried over MgSO4, filtered, and concentrated under vacuum. The crude product is purified by PTLC (MeOH/CH2Cl2 = 1 :9) to afford the title compound as a clear oil. LCMS (Method 3) m/z (M+H): 431.22, TR = 2.38 min.
122. 3,5-DΪMETHOXY-N-[8-(4-METHOXY-2,6-DlMETHYLBENZYL)-8- AZABICYCLOp^. lJOCTVϊ-YIJBENZAMIDE

123. N-[8-(4-METHOX Y-2,6-DIMETHYLBENZYL)-8-AZABICYCLO[3.2.1 ]OCT-3 -YL]-2-[4- ( 1 H-TETRAZOL-I -YL)PHENYL]ACETAM1DE

The title compound is prepared from 8-(4-methoxy-2.6-dimethy!benzyl)-8- azabicyclo[3.2.1]octan-3-amine and [4-(l H-tetrazol-l-yl)ρhenyl]acetic acid using the previousiy described HBTU coupling procedure. LCMS (Method 3) m/z (M+H): 461.33, TR = 1.99 min.
124. 2-(2,5-DlMETHYL-l ,3-THIAZOL-4-YL)-N-[8-(4-METHOXY-2,6-DIMETHYLBENZYL)-8- AZABICYCLO[3.2. I ]OCT-3-YL]ACETAMIDE
V N== /
 w W Y, S v OO N
w W Y, S v OO N
The title compound is prepared from 8-(4-methoxy-2,6-dimethyIbenzyi)-8- azabicycio[3.2.1]octan-3-amine and (2,5-dimethyI-l,3-thiazol-4-yl)acetic acid using the previously described HBTU coupling procedure. LCMS (Method 3) m/z (M+H): 428.27, TR = 2.10 min.
125. 3-FLUORO-N-[8-(3-METHOXYBENZYL)-8-AZABICYCLO[3.2.] ]OCT-3-YL]BENZAMIDE

Step 1. N-tδ-benzyl-S-azabicyclop^.ljoct-S-yO^-fluorobenzamide

The title compound is prepared from 8-benzyl-8-azabicyclo[3.2. I]octan-3-amine and 3- fluorobenzoyl chloride using the previously described Schotten-Baumann amidation procedure. LCMS (Method 3) m/z (M+H): 339.17, TR = 2.06 min.
Step 2. N-8-azabicyclo[3.2.1]oct-3-y!-3-fluorobenzainide
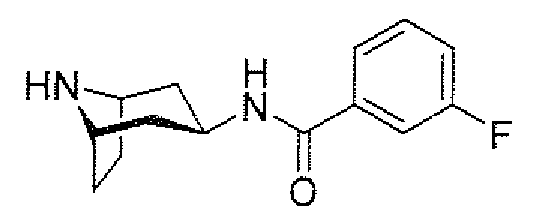
A mixture of N-fS-beiizyl-S-azabicycloP^. lJoct-j-yl^-fluσrobenzamide (7.5 g, 22.2 mmol) and 20% Pd(OH)2 on carbon (1 g) is suspended in 80 mL MeOH. The resulting mixture is hydrogenated at 60 psi H2 for 18 h. The reaction mixture is filtered through Celite® to remove the Pd catalyst and concentrated under vacuum to afford the title compound as a yellow solid. The crude product is carried forward as is. LCMS (Method 3) m/z (M+H): 249.14, TR = 1.67 min.
Step 3. 3-fluoro-N-[8-(3-melhoxybenzyl)-8-azabicyclo[3.2. ]]oct-3-yi]benzamide

The following illustrates a general procedure for the reductive aminatioπ of an amine with an aldehyde using sodium triacetoxyborohydride in the presence of acetic acid, which is referred to elsewhere herein as Procedure II.
To a screw-capped vial is added 0.2 M N-S-azabicyclofS^.lJoct-S-yl-θ-fluorobenzamide in toluene (0.2 rnL, 0.04 rnmol), 0.2 M 3-methoxybenazaldehyde in toluene (0.24 mL, 0,048 mmol), 0.2M NaBH(OAc)3 in toluene (1.0 mL, 0.2 mmol), and 0.2 M AcOH in toluene (0.3 mL, 0.06 mmol). The resulting mixture is sealed and heated at 50 0C for 18 h. The reaction mixture is quenched with I mL saturated aqueous NaHCO3, stirred for 5 min, and then the organic phase is removed and deposited directly onto an SCX column. Elution with 5 mL EtOAc (to waste) followed by elution of the product with EtOAc/TEA/MeOH 8: 1 : 1 and subsequent evaporation under vacuum affords the title compound as a clear oil. LCMS (Method 3) m/z (M+H): 369.14, TR = 2.18 min.
126. 3 -FLUORO-N-[S-O-METHOXY^-METHYLBENZYL)-S-AZABICYCLO[S .2. l]OCT-3- YL]BEN2AM1DE
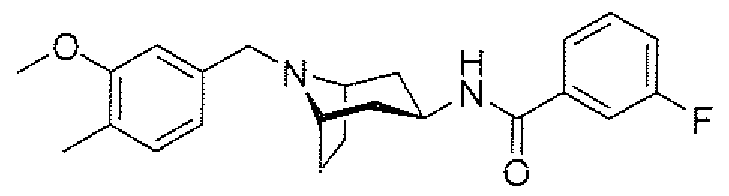
The title compound is prepared from N-8-azabicycJo[3.2.1]ocl-3-yl-3-fluorobenzamide and 3-methoxy-4-methyIbenzaldehyde using the reductive aminatioπ procedure (Procedure II) described above. LCMS (Method 3) m/z (M+H): 383.16, TR = 2.39 min.
127. 3-FLUORO-N-[8-(3-METHOXY-2-METHYLBENZYL)-8-AZABlCYCLO[3.2.1 ]OCT-3- YL]BENZAMIDE

The title compound is prepared from N-8-azabicyclo[3.2.1]oct-3-yl-3-fluorobenzamide and 3-methoxy-2-methylbenzaldehyde using the reductive amination procedure (Procedure II) described above. LCMS (Method 3) m/z (M+H): 383.17, TR = 2.23 min.
128. 3 -FLUORO-N- [8-( /H-INDOL-5 -YLMETHYL)- 8- AZAB ICYCLO[S .2.1] OCT-3 - YL]BENZAMIDE
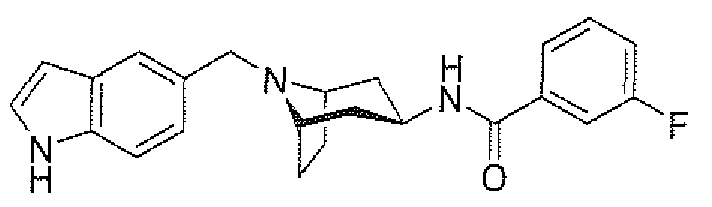
The title compound is prepared from N-8-azabicyclo[3.2.1]oct-3-yl-3-fluorobenzamide and /H-indole-5-carba3dehyde using the reductive animation procedure (Procedure II) described above, except that the amine is added as a 0.3 M solution in DMF. LCMS (Method 3) m/z (M+Η): 378.16, TR = 2.10 min.
129. N-tδ-^-CYANO-S-METΗOXYBENZYLΗ-AZABICYCLOp .2. l]OCT-3-YL]-3- FLUOROBENZAMIDE Zn(CN)2, Pd2dba3 dppf, DMF, 8O0C
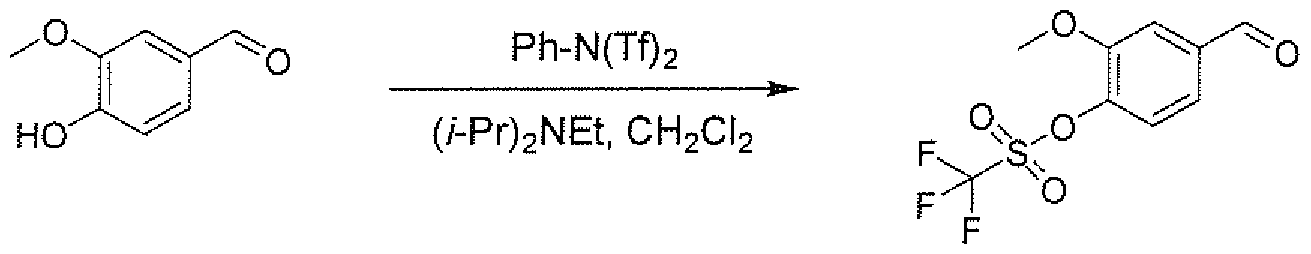
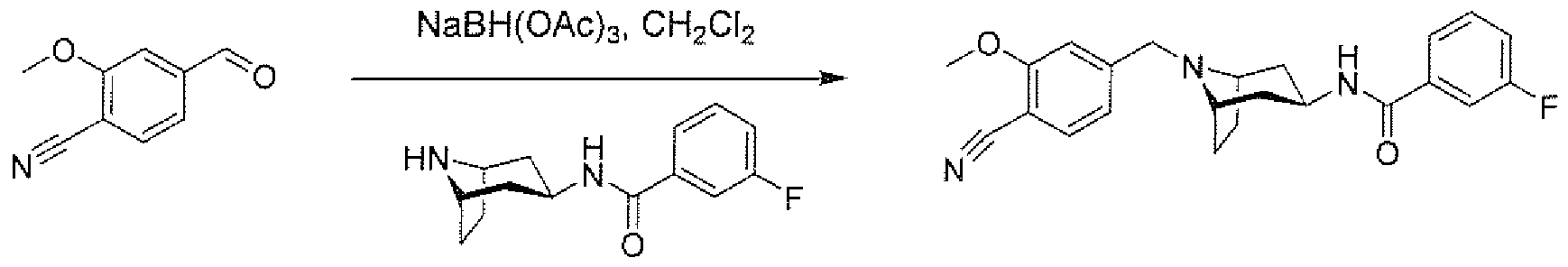
Step 1. 4-formy!-2-methoxyρhenyI trifluoromethanesulfonate

To a solution of 4-hydroxy-3-methoxybenzaldehyde (2.3 g, 15.3 mmol) and N5N- diisopropyielhylamine in 80 mL CH2CI2 is added N-phenyl-bis(trifiuoromethanesulfonimide) (7.0 g, 19.6 mmol). The resulting mixture is stirred at rt for 30 min. The reaction mixture is washed with IN HCi. brine, dried over MgSO4. filtered, and concentrated under vacuum. The crude product is purified by flash silica gei chromatography (Hex/EtOAc = 3: 1 ) to afford the title compound as a clear oil.
Step 2. 4-formyl-2-methoxybenzonitrile
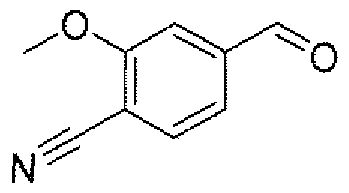
To a solution of 4-formyl-2-methoxyphenyl trifluoromethanesulfonate (1.8 g, 6.33 mmol) in 15 mL DMF is added zinc cyanide (3.7 g, 31.6 mmol). Pd2dba3 (580 mg, 0.63 mmol), and DPPF
(350 mg, 0.63 mmol). The resulting mixture is heated at 80 0C for 18 h. The reaction mixture is cooled to rt, diluted with EtOAc, washed with saturated aqueous NaHCO3, brine, dried over MgSO4, filtered, and concentrated under vacuum. The crude product is purified by flash siiica gel chromatography (Hex/EtOAc = 3: 1) to afford the titie compound as a light yellow solid. Step 3. N-[8-(4-cyano-3-methoxybenzyl)-8-azabicyclo[3.2.1]oct-3-yl]-3-fluorobenzamide
The title compound is prepared from re/-N-[(lR,3S)-8-azabicyclo[3.2.1]oct-3-yI]-3- fluorobenzamide and 4-formyl-2-methoxybenzonitrile using the reductive animation procedure (Procedure I) described above. LCMS (Method 3) m/z (M+H): 394.15, TR = 2.08 min.
130. N-(3-CHLORO-4-FLUOROPHENYL)-8-(4-METHOXY-2,3-DIMETHYLBENZYL)-8- AZABICYCLO[S


Pd(OAc)2, dppf, CO
MeOH, TEA, DMF


Step 1. S-^-methoxy^j-dimethylbenzy^-S-azabicycloP^. ljoctan-S-one

The title compound is prepared from 8-azabicycio[3.2.1]octan-3-one and 4- methoxy-2,3-dimethylbenzaldehyde using the previously described reductive amination procedure (Procedure I). LCMS (Method 3) m/z (M+H): 274.06, TR = 2.16 min.
Step 2. 8-(4-methoxy-2,3-dimethylben2yl)-8-azabicyclo[3.2, l]oct-2-en-3-yl trifluoromethanesulfonate

Step 3. Methyl 8-(4-methoxy-2,3-dimethylbenzyl)-8-azabicyclo[3.2.1]oct-2-ene-3-carboxylate

A mixture of 8-(4-methoxy-2,3-dimethyIbenzyl)-8-azabicyclo[3.2.1]oct-2-en-3-yl trifluoromethanesulfonate (3.3 g, 8.1 mmol), TEA (2.27 mL, 16.2 mmol), palladium acetate (55 mg. 0.24 mmol), and DPPF (270 mg, 0.48 mmol) are combined in a mixture of 12 mL MeOH and 8 mL DMF. The resulting mixture is stirred under a balloon of carbon monoxide for 18 h. The reaction mixture is concentrated under vacuum, diluted with EtOAc, washed with H2O, brine, dried over MgSO4, filtered, and the solvents are removed under vacuum. The crude product is purified by flash silica gel chromatography (Hex/EtOAc = 4: 1) to afford the title compound as a yellow oil. LCMS (Method 3) m/z (M+H): 316.05, TR = 2.08 min.
Step 4, Methyl 8-(4-methoxy-2,3-dimethylbenzyl)-8-azabicyclo[3.2.1]octane-3-carboxylate

A mixture of S-^-methoxy^S^limethylbenzyO-S-azabicyclop^.lJoct^-ene-θ- carboxylate (3.3 g, J 0.5 mmoi) and 10% Pd on carbon (450 mg) are suspended in 100 mL EtOAc, and the resulting mixture is hydrogenated at 40 psi H2 for 18 h. The reaction mixture is filtered through Celite® to remove the Pd catalyst and the solvent is removed under vacuum to afford the title compound as very viscous yellow oil. LCMS (Method 3) m/z (M+H): 318.08, TR = 2.05 min.
Step 5. 8-(4-methoxy-2,3-dimethyIbenzyl)-8-azabicycIo[3.2.ϊ]octane-3-carboxyltc acid

To a mixture of methyl 8-(4-methoxy-2,3-dimethylbenzyl)-8-azabicyclo[3.2.I ]octane-3- carboxylate (290 mg, 0.9 mmol) suspended in 5 mL H2O is added 0.5 mL concentrated HCi. The resulting mixture is heated at reflux for 18 h. The reaction mixture is cooled to it, and the solvent is
removed under vacuum to give the title compound as its hydrochloride salt (yellow solid). The crude product is carried forward as is.
Step 6. N-(3-chloiO-4-fluorophenyl)-8-(4-methoxy-2,3-dimethylbenzyl)-8-azabicyclo[3.2. I]octane- 3-carboxamide

To a mixture of 8-(4-methoxy-2,3-dimethylbenzyI)-8-azabicyclo[3.2.1]octane-3- carboxylic acid (200 mg, 0.59 mmol), 3-ch!oro4-fluoroaniline (170 mg, 1.18 mmol), EDCI (282 mg, 1.47 mmol), and 1 -hydroxybenzotriazole hydrate (200 mg, 1.47 mmo!) in 5 mL CH2CI2 is added TEA (0.33 mL, 2.35 mmol). The resulting mixture is stirred at rt for 18 h. The reaction mixture is
10 diluted with EtOAc5 washed with saturated aqueous NaHCO3, brine, dried over MgSO4, filtered and concentrated under vacuum. Purification by PTLC (MeOH (2M NH3)ZCH2Cl2 = 5:95) affords the title compound as a clear oil. LCMS (Method 3) m/z (M+H): 431 .09, TR = 2.63 min.
131. 3-CHLORO-4-FLUORO-N-{8-[(/5)-l-(4-METHOXY-2,3-DIMETHYLPHENYL)ETHYL]- 8-A2ABICYCLO[3.2.1 ]OCT-3-YL}BENZAMlDE



Steps 1 -3. (iS)-l-(4-methoxy-2,3-dimethy!phenyi)ethanamine
1 ) (R)-{+)-2-methyl-2- propanesulfinamide
 2) MeMgCI, AIMe3
2) MeMgCI, AIMe3

3) HCI, dioxane
The title compound is prepared using the previously described procedure (see synthesis of (i?)-l-[4-((5)-I-amino-ethyl)-2,3-dimethyI-phenoxy]-propan-2-ol), with the exception
thai the sequence commences with 4-methoxy-2,3-dimethylbenzaidehyde. LCMS (Method 3) m/z (M+H): 163.22, TR = 1.97 min observed for the -NH3 fragment.
Step 4. 8-[(75)-l-(4-methoxy~2,3-dimethyiphenyl)ethyl]-8-azabicyclo[3.2. l]octan-3-one

The title compound is prepared from (5)-] -(4-methoxy-2,3-dimethylphenyl)ethanamine using the previously described procedure employed for the synthesis of the tropane core. LCMS (Method 3) m/z (M+H): 288. ] 3, TR = 2.22 min. Chiral HPLC analysis indicates an er of 94:6 (S:R).
Steps 5-6. S-K/^-l^-methoxy^^-dimethylpheny^ethyll-S-azabicyclofS^.lJoctan-S-amine

Step 7. 3-chloro-4-fluoro-N-{8-[(l S)-l-(4-methoxy-2,3-dimethyJphenyl)ethyI]-8- azabicyclo[3.2.1]oct-3-yl}benzamide

The title compound is prepared from 8-[(lS)-l -(4-methoxy-2,3-dimethylphenyl)ethyl]-8- azabicyclo[3.2.1]octan-3-amine and 4-fluoro-3-chlorobenzoyl chloride using the previously described Schotten-Baumann amidation procedure. LCMS (Method 3) m/z (M+H): 445.14, TR = 2.65 min. Chiral HPLC analysis indicates an er of 87:13 (S:R).
1-30. 8-[(7J!)-l-(4-METHOXY-2.3 -DlMETHYLPHENYL)ETHYL]-8- AZAB ICYCLO[S.2.1]OCTAN-3~ONE

The title compound is prepared from 4-methoxy-2,3-dimethylbenzaldehyde essentially as described above, with the exception that (5)-(-)-2-methyl-2-propanesulfinamide is used as the chiral auxiliai-y. LCMS (Method 3) m/z (M+H): 288.13, TR = 2.22 min. Chiral HPLC analysis indicates an er of 95: 5 CSVi?).
132. AMIDATION OF 8-[l-(4-METHOXY-2,3-DIMETHYLPHENYL)ETHYL]-8- AZABICYCLO [3.2.1 JOCTAN-3 -AMINE

The following is a genera! procedure for the synthesis of amides of such as amides of 8-[l- (4-methoxy-2,3-dimethylpheny])ethyl]-8-azabicyclo[3.2.1]octan-3-amine and related compounds (e.g., 8-[l -(4-methoxy-3-methylphenyl)ethyl]-8-azabicyclo[3.2.1]octaπ-3-amine and 8-[l -(4- methoxy-2-methy]ρhenyl)ethyi]-8-azabicyclo[3.2.1]octan-3-amlne).
The primary amine (150 μL of a 0.2N solution in toluene, 30 μmol), any desired acid (180 μL of a 0.2N solution in DMA, 36 μmol), and TEA (90 μL of a 1.0N solution in toluene, 90 μmol) are shaken for 5 min at rt before adding HBTU (225 μL of a 0.2N solution in DMA, 45 μmol). Subsequent shaking for 1 h at rt leads to complete conversion to the amide as determined by LCMS. 0.1 mL of water is added and the homogeneous mixture is evaporated at 50 0C using either IR- Dancer or Genevac. The residue is dissolved in 1 mL of EtOAc before 0.5 mL of a water/saturated potassium carbonate-mixture (4/1) is added. The organic layer is decanted and put onto a 0.5 g pre- packed column of silica based benzenesulfonic acid resin (loading ~0.9 mmol/g). The remaining aqueous layer is extracted with 2 more 1 mL portions of EtOAc which are also deposited onto the column. The loaded column is washed with EtOAc (2 mL) and 5 mL of EiOAc/MeOH (10/1) before the product is eluted with 10 mL of EtOAc/MeOH/TEA and concentrated under reduced pressure. To remove any residual TEA, the dried sample is dissolved in 10 mL of DCM and stripped of its solvent twice. Purity of the product is determined by LCMS; compounds may be further purified on silica gel using DCM/MeOH(2N NH3) in various mixtures (50/1 to 10/1).
Compounds in Table I are prepared using this procedure. In Table I, compounds with a "*" in the column headed '1K1" have a K1 in the assay of Example 10 that is less than 1 micromolar. The molecular weight (presented as M+I, determined using Method 3) is shown in the column headed "MS" in Table 1, along with the retention time in minutes.
Table I
Compound Name K1 IR MS
N-{8-[l -(4-methoxy-2,3- dimethylphenyl)ethyl]-8- azabicyclo[3.2.1 ]oct-3-yl}- * 1.23 492.24
6-(2,2.2-trifl uoroethoxy ) nicotinamide
2-methoxy-N-{8-[l-(4- methoxy-2,3- dimethy ipheny 1 )ethy 1] -8 - * 1.21 428.28 azabicyclo[3.2.1]oct-3-
 yl}nicotinamide
yl}nicotinamide
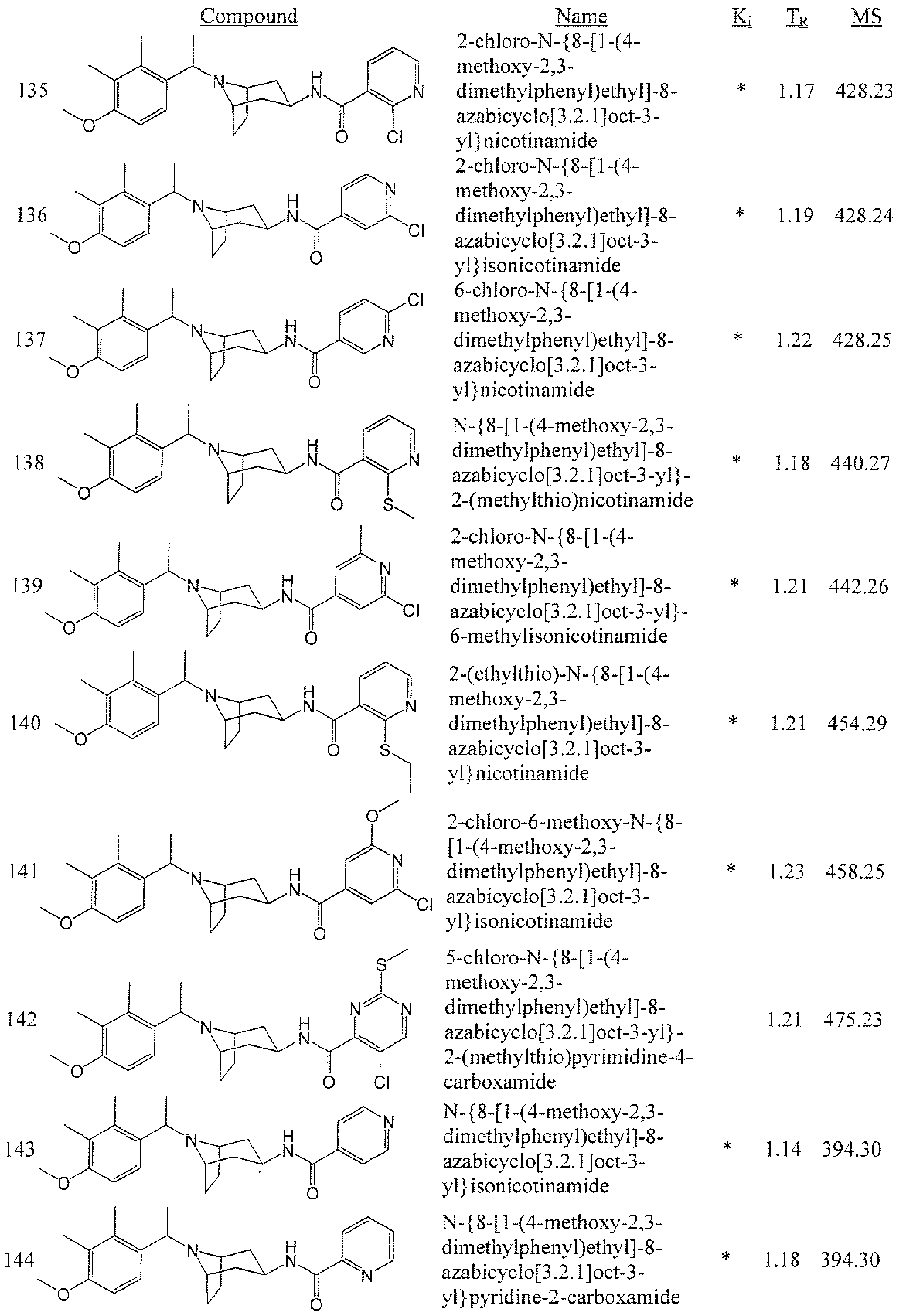
 Compound Name JC, IE MS
Compound Name JC, IE MS

-8 - 1.22 462.19 -
- 1.26 499.32

- 8 -
* 1.20 437.31 i de
* 1.20 4.3731
* 1.23 437.31
8- * 1.23 441 .25
* 1.22 441.27
* 1.2] 441.27
* 1.21 443.28
* 1.23 457.24
 Name K1 IR MS
Name K1 IR MS
2-(2,5-dimethoxypheny])- N-{8-[l-(4-methoxy-2,3- dimethylphenyl)ethyl]-8- .19 467.3 ] azabicyclo[3.2, 1 ]oct-3- y]}acetamide

2-(4-chloro-2-

2-(3-chlorophenoxy)-N-{8-
[l-(4-methoxy-2,3- dimethylphenyl)ethyl]-8- * 1.24 471.27 azabicyclo[3.2. l]oct-3- yl}propanamide
2-(3,5-difluorophenyl)-N- {8-[l-(4-methoxy-2,3- dimethylphenyl)ethyl]-8- 1.21 443.28 azabicyclo[3.2.1 ]oct~3- yl}acetamide

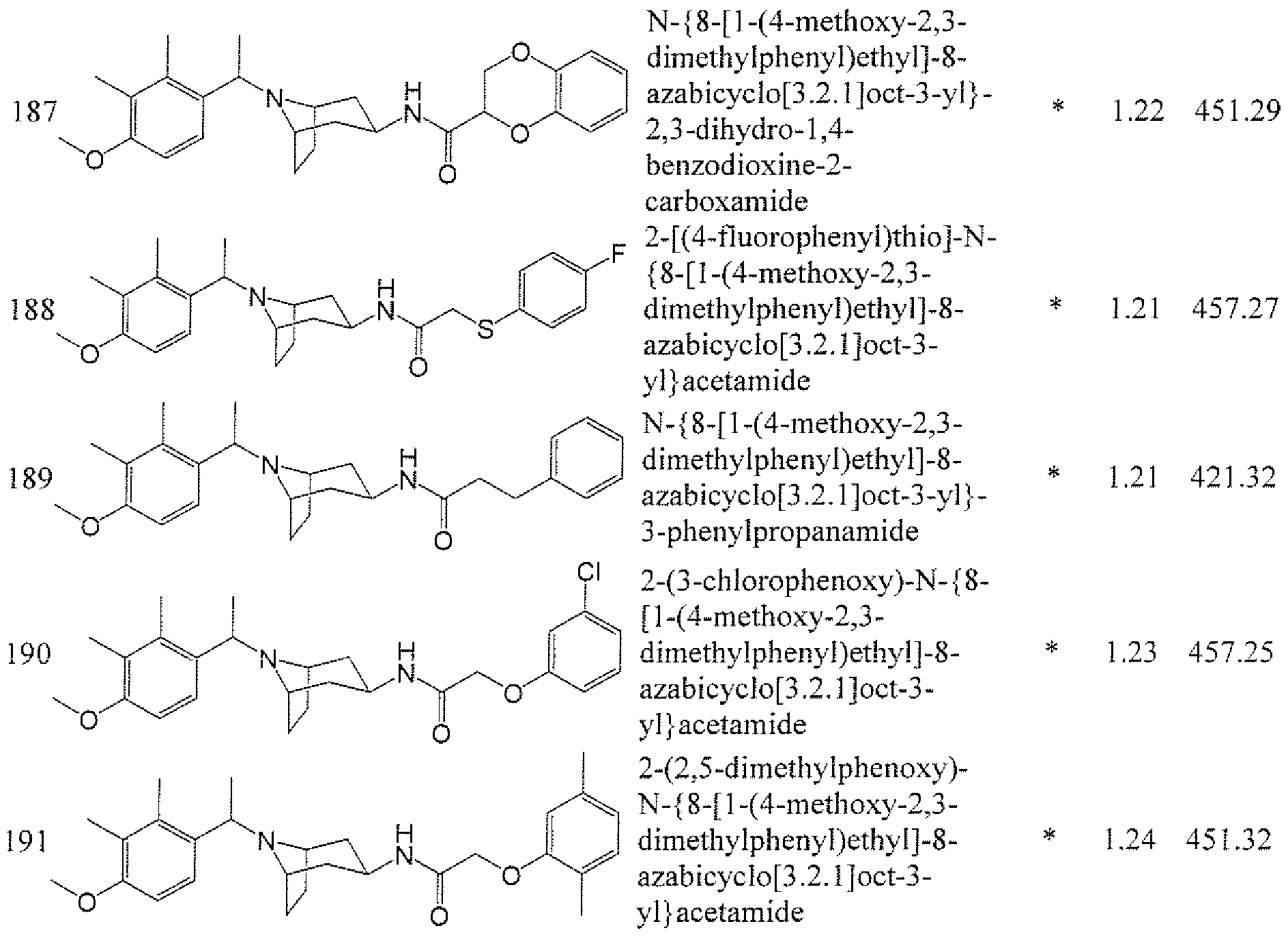
2-(3,4-difluoropheny!)-N-
{8-[I -(4-methoxy-253- dimethylphenyl)ethyl]-8- 1.21 443.29 azabicyclo[3.2.1]oct-3- yl}acetamide



 Compound Name ^ IK MS
Compound Name ^ IK MS
N-{8-[I-(4-methoxy-2.3- dimethy lph eny 1 )ethy 1] -8 - azabicyclo[3.2.1 ] oct-3 -yl}- 1.17 415.25 4-methyl- 1 ,2,3-thiadiazole-
 5-carboxamide
5-carboxamide

2-(2,5 -dimethyl- 1 ,3-thiazol-
4-yI)-N-{8-[] -(4-methoxy-
2,3-dimethylphenyl)ethyl]- 1.17 442.29
8-azabicyclo[3.2.1]oct-3-
 yl}acetamide
yl}acetamide
 Compound Name K1 MS
Compound Name K1 MS
3 -ch loro-4 - methoxy-N - { 8 -
[!-(4-methoxy-2,3- dimethy]phenyl)ethyl]-8- * 1.22 457.25 azabicyclo[3.2.1]oct-3-
 yljbenzamide
yljbenzamide
 Compound Name K1 IR MS
Compound Name K1 IR MS

2-(2,4-dichloroρhenoxy)-N- {8-[l-(4-methoxy-2,3- dimethylphenyl)etliyl]-8- 1.26 491.18 azabicyclo[3.2.1 ]oct-3- yljacetamide N-{8-[l-(4-methoxy-2,3- dimethylphenyl)ethyl]-8- azabicyclo[3.2.1]oct-3-yl}- 1.21 453.28 2-(3-methoxyphenoxy) acetamide
2-biphenyl-4-yl-N-{8-[l-(4- methoxy-2,3- dimethylphenyI)ethyl]-8- 1 .26 483.29 azabicyclo[3.2.1]oct-3- yl}acetamide
3-fluoro-N-{8-[l-(4- methoxy-2,3- dimethyIphenyl)ethyl]-8- 1.23 425.27 azabicycio[3.2. I]oct-3-yl}- 5-methyIbenzamide
3-cyano-N-{8-[l-(4- methoxy-2,3- d imethylpheny l)ethy 1] - 8- 1.18 418.25 azabicyclo[3.2.1 ]oct-3- yijbenzamide melhyl 3-({8-[l-(4- methoxy-2,3- dimethyiphenyl)ethyl]-8- 1.21 451.27 azabicyclo[3.2.1 ]oct-3-
 y 1 } c arbam oy 1 )benzoate 3-(dimetliylamino)-N-{8-[l- (4-methoxy-2,3- d imethylpheny 1 )ethy 1 ] -8 - 1.17 436.30 azabicyclo[3.2.1]oct-3- yl}benzamide 3-nitro-N-{8-[l-(4- methoxy-2,3- dimethylphenyi)ethyi]-8- 1.20 438.24 azabicyclo[3.2.1]oct-3- yl}benzamide
y 1 } c arbam oy 1 )benzoate 3-(dimetliylamino)-N-{8-[l- (4-methoxy-2,3- d imethylpheny 1 )ethy 1 ] -8 - 1.17 436.30 azabicyclo[3.2.1]oct-3- yl}benzamide 3-nitro-N-{8-[l-(4- methoxy-2,3- dimethylphenyi)ethyi]-8- 1.20 438.24 azabicyclo[3.2.1]oct-3- yl}benzamide
N-{8-[l-(4-methoxy-2,3- dimethylphenyI)ethyl]-8- azabicyclo[3.2. ljoct-3-yl } - 1.21 490.25 2-methyi-4-phenyl-l,3- thiazole-5-carboxamide

 334-dichloro-N-{8-[l-{4- methoxy-3- methylphenyl)ethyl]-8- 1 .24 447.12 azabicyclo[3.2.1 ]oct-3- yl}benzamide
334-dichloro-N-{8-[l-{4- methoxy-3- methylphenyl)ethyl]-8- 1 .24 447.12 azabicyclo[3.2.1 ]oct-3- yl}benzamide
3,5-dimethoxy-N-{8-[l-(4- methoxy-3- methylphenyl)ethy]]-8- 1.18 439.24 azabicycIo[3.2.1]oct-3-
 yl}benzamide
yl}benzamide
3-chloro-4-methoxy-N-{8- [l-(4-methoxy-3- methy lpheny l)ethy 1] - 8 - 1.20 443.17 azabicyclo[3.2.1]oct-3-
 yl}benzamide
yl}benzamide

2-chloro-N-{8-[l-(4- methoxy-3- methylphenyl)ethyϊ]-8- * 1.16 414. J 6 azabicyclo[3.2. ] ]oct-3- yl}isonicotinamide N-{8-[l-(4-methoxy-3- methyIpheπyl)ethyl]-8- azabicycio[3.2.I]oct-3- 1.15 385.19 yl}thiophene-2- carboxamide
N-{8-[l -(4-methoxy-3- methylphenyl)ethyl]-8- azabicyclo[3.2.1]oct-3- 1.15 385.18 yl}thiophene-3- carboxamide
2-chloro-6-methoxy-N- {8- [] -(4-methoxy-3- methylρhenyl)ethyl]-8- 1.21 444.10 azabicycIo[3.2.1]oct-3- yl}isonicolinamide
5-bromo-N-{8-[l-(4- methoxy-3- methylphenyI)ethy]]-8- 1.18 558.09 azabicyclo[3.2.1]oct-3- y]}nicotinamide
3 -chloro-4-fluoro-N- { 8- [ 1 - (4-methoxy-3- methylphenyl)ethyl]-8- 1.22 431.15 azabicyclo[3.2. l]oct-3- yi}benzamide
3-fluoro-N-{8-[l-(4- methoxy-3- melhylphenyl)ethyl]-8- 1.20 41 1.22 azabicyclo[3.2. I]oct-3-yl}- 5-methylbenzamide
3-cyano-N-{8-[I-(4- methoxy-3- methylphenyl)ethyl]-8- 1.16 404.21 azabicyclo[3.2.1]oct-3- yljbenzamide
N-{8-[l -(4-methoxy-3- methyIpheny])ethyl]-8- azabicyclo[3.2.1]oct-3-y!}- 1.20 431.15 5-( methy lth io )thiophene-2-
 carboxamide
Name K1 TP MS
carboxamide
Name K1 TP MS
N-{8-[I-(4-methoxy-3- methylphenyl)ethyl]-8- azabicyclo [3.2.1] oct-3 -y i } - * 1.16 394.22 6-methylpyridine-2- carboxamide 6-bromo-N-{8-[l-(4- methoxy-3- methylpheny!)ethyi]-8- 1.19 458.11 azabicyclo[3.2.1 joct-3- yl } pyridine-2-carboxam ide 5,6-dichloro-N-{8-[l-(4- methoxy-3- methylphenyl)ethyl]-8- 1.21 448.11 azabicycIo[3.2.1]oct-3- yl}nicotinamide
N-{8-[l-(4-methoxy-3- methylphenyl)ethyl]-8- 1 .20 407.25 azabicyclo[3.2.1]oct-3-yl}- 3,4-dimethyIbenzamide
N-{8-[l-(4-methoxy-3- methylphenyl)ethyl]-8- 1.21 407.26 azabicycio[3.2.1]oct-3-yl}- 3,5-dimethylbenzamide
N-{8-[l-(4-methoxy-3- methylphenyl)ethyl]-8- azabicyclo[3.2. i]oct-3-yl} - 18 449.19 2-(trifluoromethyl) pyrimidine-5-carboxamide
N-{8-[l -(4-methoxy-3- methy]phenyl)ethyi]-8- 1.19 393.24 azabicyc!o[3.2.1 ]oct-3-yl} - 3-methyIbenzamide
2-(3,4-difluoiOphenyl)-N- {8-[l -(4-methoxy-3- methylphenyl)ethyl]-8- 1.19 429.21 azabicyclo[3.2.1 ]oct-3 - yljacetamide
2-(2,5-dimethylphenoxy)-
N-{8-[l-(4-methoxy-3- methylphenyl)ethyl]-8- * 1.23 437.26 azabicyclo[3.2.1]oct-3- yl}acetamide
N-{8-[l"(4-methoxy-3- methylphenyl)ethyl]-8- * 1 .10 394.23 azabicyclo[3.2.1] oct-3 -yl}-
 5-methylnicotinamide
Name K, XR MS
5-methylnicotinamide
Name K, XR MS
3-fϊuoro-N-{8-[l-(4- methoxy-2- methylphenyl)ethyl]-8- 1.15 397.21 azabi eye Io [3.2.1 ] oct-3 - yljbenzamide 3-methoxy-N-{8-[l-(4- methoxy-2- methylphenyl)ethy!]-8- * 1.15 409.23 azabicyclo[3.2.1] oct-3 - yl}benzamide 3-fluoro-N-{8-[l-(4- methoxy-2- methylpheny])ethyl]-8- 1.19 41 1.22 azabi cyclo[3.2.1] oct-3 -yl } - 4-methyIbenzamide 3-chloro-N-{8-[I-(4- methoxy-2- methylphenyl)ethyl]-8- 1.18 413.17 azabicyclo[3.2.1]oct-3- yljbenzamide 3,4-difluoro-N-{8-[l-(4- methoxy-2- methylphenyI)ethyl]-8- 1.17 415.20 azabicyclo[3.2.1]oct-3- yi}benzamide
3-ethoxy-N-{8-[I -(4- methoxy-2- methylphenyl)ethyl]-8- 1 .18 423.24 azabicyc!o[3.2. I]oct-3- yl}benzamide
3,5-dichloro-N-{8-[l-(4- methoxy-2- methy]phenyl)ethyl]-8- 1.23 447.13 azabicyclo[3.2.1] oct-3 - y]}benzamide 3,4-dichloro-N-{8-[l -(4- methoxy-2- methy!phenyl)ethyl]-8- 1.23 447.12 azabicyclo[3.2. l]oct-3- y!}benzamide
3s5-dimelhoxy-N-{8-[1 -(4- methoxy-2- methy!phenyl)ethyl]-8- 1.17 439.24 azabicyc!o[3.2. I]oct-3- yl}benzamide
3-chloro-4-methoxy-N-{8-
[l-(4-methoxy-2- methylphenyl)ethyI]-8- * 1.18 443.17 azabicyclo[3.2.1]oct-3-
 yl}benzamide
Compound Name K1 Ts MS
yl}benzamide
Compound Name K1 Ts MS
N-{8-[l-(4-methoxy-2- methyIphenyI)ethyl]-8- azabicyclo[3.2.1]oct-3-yl}- 1.20 447.19 3 -(trifluoromethyl) benzamide
N-{8-[l-(4-methoxy-2- methylphenyl)ethyl]-8- 1.18 397.24 azabicydo[3.2.1]oct-3-yl}- 2,5-dimethyl-3-furamide
N-{8-[l-(4-methoxy-2- methy]phenyl)ethyI]-8- 1.15 383.22 azabicyclo[3.2.1 ]oct-3-yl} - 2-methyl-3-furamide NN--{{88--[[ll--((44--mmeetthhooxxyy--22-- methy lpheny 1 )ethy I]-- 88 -- 1.1 1 369.23 azabicyclo[3.2.1 ]oct--33--;yl}-
3-furamide
N-{8-[l-(4-methoxy-2- methylphenyi)ethyl]-8- 1.16 397.24 azabicyclo[3.2.1]oct-3-yI}- 4.5-dimethyI-2-furamide
3-chloro-N-{8-[l-(4- methoxy-2- methy!phenyI)ethyl]-8- 1.21 427.20 azabicycio[3.2, 1 ]oct-3-yl} -
4-methyibenzamide
2-chloro-N-{8-[l-(4- methoxy-2- methylphenyl)ethyl]-8- 1.14 414.16 azabicyclo[3.2.1 ]oct-3- yl}isonicotinamide
N-{8-[l-(4-methoxy-2- methylphenyl)elhyl]-8- azabicyclo[3.2. l]oct-3 - 1.13 385.18 yi}thiophene-2- carboxamide
N~{8~[l-(4-methoxy-2- methylphenyI)ethyl]-8- azabicyclo[3.2.1 ]oct-3- * 1 .13 385.18 yl}thiophene-3- carboxamide
2-chioro-6-methoxy-N-{8- [l -(4-methoxy-2- methylphenyl)ethyl]-8- * 1.20 444.18 azabicyclo[3.2.1 ]oct-3-
 yl} isonicotinamide
Name £ IR MS
yl} isonicotinamide
Name £ IR MS
2-(2,5-dimethoxyρhenyl)- N-{8-[l-(4-methoxy-2- methy ! pheny !)ethy 1] -8 - * 1.35 453.25 azabicyclo[3.2.1 ]oct-3- y]}acetamide
5-bromo-N-{8-[l -(4- methoxy-2- methy lpheny l)ethy I]- 8 - 1.15 458.1 1 azabicyc!o[3.2.1]oct-3- yl} nicotinamide
N-{8-[l-(4-methoxy-2- methy 1 pheny l)ethy 1] - 8 - 1.15 371.26 azabicyclo[3.2. l]oct-3-yl} cyclopentanecarboxamide 3-chloro-4-fluoro-N- (S-[I -
(4-methoxy~2- methylphenyl)ethyl]-8- 1.20 431.16 azabicyclo[3.2.1 ]oct-3- yl} benzamide
3-fluoro-N-{8-[l-(4- methoxy-2- methyIphenyl)ethyl]-8- 1.18 41 1.23 azabicyclo[3.2.1 ]oct-3 -yl } - 5 -methyl benzamide
3-cyano-N-{8-[l-(4- methoxy-2- methylphenyl)ethyl]-8- * 1.13 404.22 azabicycio[3.2.1 ]oct-3- yl} benzamide
N-{8-[] -(4-methoxy-2- methylphenyl)ethyI]-8- azabicyclo[3.2.1 ]oct-3-yl} - * 1.18 431.16 5-(methy lth io)thioph ene-2 - carboxamide
N-{8-[l-(4-methoxy-2- methylphenyl)ethyl]-8- azabicyclo[3.2.1]oct-3-yl}- * 1.15 394.23
6-methy!pyridine-2- carboxamide
6-bromo-N-{8-[l-(4- methoxy-2- methy lpheny l)ethy 1] -8 - 1.16 458.1 1 azabicyclo[3.2. l]oct-3-
 yI}pyridine-2-carboxamide
yI}pyridine-2-carboxamide

346. PREPARATION OF UREAS AND SULFONAMIDES FROM 8-[ l -(4-METHOXY-2,3- DIMETHYLPHENYL)ETHYL]-8-AZABICYCLO[3.2. 1 ] OCT AN-3 -AMINE AND RELATED COMPOUNDS

347. 3-CHLORO-N-(8-{ l -[2,3-DIMETHYL-4-(MORPHOLINE-2-YLMETHOXY)PHENYL]ETHYL}-8- AZABICYCLO[3.2.1 ]OCT-3-YL)-4-FLUOROBENZAMIDE

Step I . Morpholin-2-yImethanoi Commercially available 3-amino-l,2-propandiol is converted to morpholine-2-yImethanol essentially as described in the literature (Organic Lett. (2005) 937+ and supplement material) except that the reaction product is not isolated but used crude in the next step after removal of most of the solvent.
Step 2. /err-Butyl-2-(hydroxymethyl)morpholine-4-carboxyIate The crude mixture from Step 1 (100% ~20 mmol) is suspended in 30 mL of water and 50 mL of DCM. After addition of 10 mL saturated sodium bicarbonate solution which adjusts the pH to 12-13, 2 equivalents of Boc-anhydrsde are added and the reaction mixture is stirred for 20 h at rt
or until LCMS shows full conversion. Subsequently, the mixture is put into 300 mL of water and extracted with DCM (3 x 200 mL). Diying over magnesium sulfate and evaporation of the solvent yields a crude material that is purified on siiica gel using straight EtOAc. LCMS (Method 3) m/z (M+Na):240.01. Step 3. før/-Butyl 2-({[(4-methylphenyl)suifonyl]oxy}methyl)moφholine-4-carboxylate
The purified aicohol from Step 2 (1.81 g. 8.33 mmol) is dissolved in 20 mL of DCE. Addition of TEA (10.5 mL, 75.0 mmol) and DMAP (101 mg, 0.83 mmol) is followed by cooling to 0 0C. After addition of tosyl chloride (1.75 g, 9.16 mmol), the reaction mixture is allowed to warm to rt. Subsequently, the solution is transferred into 200 mL of water, basified with saturated potassium carbonate (10 mL), extracted with DCM (3 x 100 mL), and dried over magnesium sulfate. Final purification in silica gel using acetyl acetate/DC M/hexane (1/1/2) yields the title compound. LCMS (Method 3) m/z (M+Na):394.09.
Step 4. /err-Buty!-2-{[4-(l-{3-[(3-chloro-4-fluorobenzoyl)amino]-8-azabicyclo[3.2.1]oct-8- y 1 } ethy i)-2 ,3 -d im eth y 1 ph enoxy] methyl } morpho 1 ine-4-carboxy late The tosylate from Step 3 (129 mg, 348 μmol), previously described phenol (100 mg, 232 μmol). and /eϊra-butylammonium iodide (86 mg, 232 μmol) are dissolved in 5 mL of DMF. After addition of NaH (60% in mm. oil, 37 mg, 928 μmol), the reaction mixture is stirred for 20 h at rt before being put into 150 mL of Et2O and EtOAc (1 : 1). The organic layer is washed with water/saturated potassium carbonate (100 mL + 10 mL) and water (2 x 100 mL) and then dried over magnesium sulfate. After removal of the solvent, the crude material is subjected to a catch-release chromatography (2.0 g S1O2 based benzenesulfonic acid resin, 0.7-0.9 mmol/g loading). The compound is put onto the column with EtOAc (3 x 2 mL), then washed with EtOAc (4 mL) and EtOAc/MeOH (20/1 , 10 mL) before being eluted with EtOAc/MEOH/TEA (10/1/1, 20 mL). Final purification on silica gel with DCM/MeOH(2M NH3) (15/1) yields the title compound. LCMS (Method 3) m/z (M+H):630.2S.
Step 5. 3-Chloro-N-(8-{ 1 -[2,3-dimethyl-4-(moψholin-2-ylmethoxy)phenyl]ethyl}-8-azabicyc]o [3.2.1]oct-3-yl)-4-fluorobenzamide
The Boc-protected compound from Step 4 (81 mg, 129 μmol) is dissolved in 5 mL of DCM and 2 mL of MeOH. Addition of 4N HCL/dioxane (5 mL) and stirring at rt for 2.5 h results in complete Boc removal as validated by LCMS. Subsequently, the reaction mixture is put into 100 mL of water, basified with saturated potassium carbonate (10 mL), extracted with DCM (3 x 100 mL), and dried over magnesium sulfate. Final purification on silica gel with DCM/MeOH(2N NH3) (10/1) yields the title compound. LCMS (Method 3) nι/z (M+H):530.18.
348. 3-CHLORO-N-(S-( I-P5B-DIMETHYL^-(MORPHOIJNE-Z-YLMETHOXY)PHENYL]ETHYL) -S- AZABICYCLO[3.2.1 ]θCT-3 -YL) -4-FLUOR0BENZ AMIDE

The preparation of the (i?)-morpho!ine compound follows the previously outlined synthesis of the (^-stereoisomer starting with the commercially available (Λ)-3-amino- I ,2-propanediole.
349. 3-CHLORO-N-[8-(1 -{2J-DIMETHYL-4-[(5-OXOMORPHOLJN-2-YL)METHOXY]PHENYL} ETHYL)- 8-AZABiCYCLO[3.2.1]OCT-3-YL]-4-FLUOROBENZAMIDE

Step ] . 6-(Hydroxγmethyl)morpholin-3-one Commercially available 3-amino-l,2-propandiol is converted to 6-
(hydroxymethy])mθφholin-3-oπe essentially as described in the literature (Organic Lett., 2005, 937+ and supplement material).
Step 2. (5-Oxomoipholin-2-yl)methyl 4-methylbenzenesuifonate
The purified alcohol from Step 1 (500 mg, 3.81 mmoi) is dissolved in 20 rnL of DCM. Addition of TEA (4.78 mL, 38.1 mmol) and DMAP (47 mg, 0.38 mmol) is followed by cooling to 0 0C. After addition of tosyl chloride (800 mg, 4.19 mmol), the reaction mixture is allowed to warm to rt. Subsequently, the solution is put into 100 mL of water, basified with saturated potassium carbonate (10 mL), extracted with DCM (3 x 100 mL), and dried over magnesium sulfate. Final purification on silica gel using DCM/MeOH (15/1) yields the title compound. LCMS (Method 3) m/r (M+H):285.93.
Step 3. 3-Chloro-N-[8-(l-{2,3-dimethyl-4-[(5-oxomorpholin-2-yl)methoxy]phenyl} ethy 1 )-8 -azabicyc Io [3.2.1] oct-3 -y l]-4-fluorobenzamide
The purified tosylate from Step 2 (50 mg, 174 μmol), previously described phenol (50 mg, l lόμmol). and te/rø-butyl ammonium iodide (43 mg. 1 16 μmol) are dissolved in 5 mL of DMF. After addition of NaH (60% in min. oil, 18.6 mg, 464 μmol), the reaction mixture is stirred at rt for
72 h before being put into 150 mL of EtjO and EtOAc (1 : 1 ). The organic layer is washed with
water/saturated potassium carbonate (100 mL + 10 mL), water (2 x 100 mL), and then dried over magnesium sulfate. After removal of the solvent, the crude material is subjected to a catch-release chromatography (1.0 g S1O2 based benzene sulfonic acid resin, 0.7-0.9 mmol/g loading). The compound is put onto the column with EtOAc (3 x 2 mL), then washed with EtOAc (2 mL) and EtOAc/MeOH (20/1, 8 mL) before being eluted with EtOAc/MEOH/TEA (10/1/1 , 20 mL). Final purification on silica gel with EtOAc/MeOH/TEA (10/1/1) yields the title compound. LCMS (Method 3) m/z (M+H):544.16.
350. 3-CHLORO-N-[8-( 1 - {2,3-DIMETHYL-4-[(5-OXOMORPHOLIN-2-YL)METOOXY]PHENYL}ETHYL> 8-AZABICYCLO[3.2. L ]OCT-3-YL]-4-FLUOROBENZAMIDE

The preparation of the (i?)-morpholinone compound follows the previously outlined synthesis of the (^-stereoisomer starting with the commercially available (R)-3 -amino- 1,2- propanediol.
351. 3-CHLORO-N-[8-(l-{2,3-DIMETHYL-4-[(4-METHYL-5-OXOMORPHLIN-2- YL)METHOXY]PHENYLjETHYL)-S-AZABiCYCLOP^. l]OCT-3-YL]-4-FLUOROBENZAMIDE

Step 1. (4-Methyl-5-oxomorpholin-2-yl)methyl 4-methylbenzenesuIfonate
The previously described tosylate (200 mg, 701 μmol) is dissolved in 10 mL of DMF.
Addition of methyl iodide (88 μL, 1.4 mmol) and NaH (60% in min. oil, 33mg, 825 μmol) yields the N- methylated compound after 1 h at rt. The mixture is put into 200 mL of EtOAc/ethyf ether (1/1), washed with water (2 x 200 mL), and dried over magnesium sulfate. Purification on silica gel with
DCM/MeOH (20/1 ) yields the title compound. LCMS (Method 3) m/z (M+H): 300.01.
Step 2. 3-ChIoro-N-[8-(l -{2,3-dimethyl-4-[(4-methyl-5-oxomorpholin-2-yl)methoxy]pheny!} ethyl)-8-azabicyclo[3.2.1 ]oct-3 -yl]-4-fluorobenzamide The purified N-methylated compound (80 mg, 267 μmoi), previously described phenol (77 mg, 178 μmol), /e/ra-butylammonium iodide (66 mg, 178 μmol), and cesium carbonate (174 mg, 534 μmol) are dissolved in 3 mL of DMF and heated to 80 0C overnight. Subsequently, the reaction mixture is put into 1 10 mL of water/saturated potassium carbonate and extracted with EtOAc (3 x
100 mL). After drying over magnesium sulfate, final purification on silica gel with DCM/MeOH(2N NH3) ( 15/1 ) yields the title compound. LCMS (Method 3) m/z (M+H): 558.2 ϊ .
352. 3-CHLORO-N-[S-(I -{2,3-DIMETHYL-4-[2-(5-OXOMORPHOLIN-2-YL)ETHOXY]PHENYL} ETHYL)- δ-AZABICYCLOp ^. l]OCT-3-YL]-4-FLUOROBENZAMIDE

Step 1. Methyl 4-[(chloroacetyl)amino]-3-hydroxybutanoate
Commercially available 4-amino-3-hydroxybutanoic acid (6.0 g, 50.2 mmol) is first dissolved in 200 mL of MeOH and then cooled to 0 0C. Slow addition of acetyl chloride (17.8 mL, 250 mmol) over 10 min is followed by stirring at it for 2h before the reaction mixture is evaporated. The residue is dissolved in 20 mL of MeOH and again evaporated. Subsequently, the crude mixture is dissolved in 200 mL of acetonitrile and 35 mL of MeOH. Addition of TEA (21 mL, 151 mmol) and cooling to -I O 0C is followed by slow addition of chloroacetyl chloride (4.4 mL, 55.3 mmol). The reaction mixture is allowed to warm to it overnight, evaporated under reduced pressure, and the residue is put onto 100 mL of EtOAc/MeOH (92/8). The precipitated TEAxHCl is filtered off and washed with little of the same solvent. After evaporating the solvent, the crude mixture is directly purified on silica gel using EtOAc/MeOH (95/5). LCMS (Method 3) m/z (M+H):210. I4.
Step 2. 2-ChlθH>N-(2,4-dihydroxybutyl)acetamide
The purified methyl ester from Step 1 (5.34 g, 25.4 mmol) is dissolved in 100 mL of THF and cooled to O0C. Slow addition of LiBHVTHF (2.0N, 12.7 mL, 25.4 mmol) and stirring for 90 min at the same temperature led to complete reduction of the ester. HCl/dioxane (4.0N, 6.99 mL, 27.9 mmol) is added to quench the reaction before all solvents are evaporated under reduced pressure. The resulting white/slimy residue is dissolved in 50 mL of MeOH and again evaporated to dryness. Repeating the treatment of the residue for two more times led finally to an almost clear paste that is directly purified on silica gel with EtOAc/MeOH (5/1). LCMS (Method 3) m/z (M+H): 182.12.
Step 3. 6-(2-HydroxyethyI)morpholin-3-one
The purified diol from Step 2 (575 mg, 3.16 mmol) is dissolved in 10 mL of J-amyl alcohol and slowly added to a solution of potassium tert-butoxide (890 mg, 7.92 mmol) in 20 mL of t-amy] alcohol. After 90 min, 5.5 mL MeOH/water (10/1) are added and the stirring continued for another
90 min. Subsequently, the solvent is evaporated under reduced pressure and the yellowish paste purified directly on silica gel with EtOAc/MeOH (4/1). LCMS (Method 3) m/z (M+H): 146.21.
Step 4. 2-(5-Oxomorpholin-2-yl)ethyl 4-methylbenzenesulfonate
The purified morpholinone from Step 3 (289 mg, 2.0 mmol) is dissolved in 10 mL of DCE. Addition of TEA (2.51 mL, 18 mmol). DMAP (24 mg, 0.2 mmol) and cooling to 0 0C is followed by addition of tosyl chloride (419 mg, 2.20 mmol). The reaction mixture is allowed to warm to rt over 3.5 h. Subsequently, the solution is put into 200 mL of water, basified with saturated potassium carbonate (10 mL), extracted with DCM (3 x 100 mL), and dried over magnesium sulfate. Final purification on silica gel using DCM/MeOH (20/1) yields the title compound. LCMS (Method 3) m/z (M+H):299.98.
Step 5. 3-Chloro-N-[8-(l-{2,3-dimethyl-4-[2-(5-oxomorpholin-2-yl)ethoxy]phenyl}ethyl)-8- azabicyclo[3.2.1 ]oct-3 -yl]-4-fluorobenzamide
The purified tosylate from Step 4 (52 mg, 174 μmol), previously described phenol (50 mg, 1 16 μmol), and /e/ra-butylammonium iodide (43mg, 1 16 μmol) are dissolved in 5 mL of DMF. After addition of NaH (60% in min. oil, 18.6 mg, 464 μmol), the reaction mixture is stirred at rt for 72 h before being put into 150 mL Of Et2O and EtOAc (1 :1). The organic layer is washed with water/saturated potassium carbonate (100 mL + 10 mL). and water (2 x 100 mL), and then dried over magnesium sulfate. After being stripped of the solvent, the crude material is subjected to a catch-release chromatography (1 .0 g SiO2 based benzenesulfonic acid resin, 0.7-0.9 mmol/g loading). The compound is put onto the column with EtOAc (3 x 2 mL), then washed with EtOAc (2 mL) and EtOAc/MeOH (20/1, 8 mL) before being eluted with EtOAc/MEOH/TEA (10/1/1, 20 mL). Final purification on silica gel with DCM/MeOH(2N NH3) (15/1) yields the title compound. LCMS (Method 3) m/z (M+H): 558.25.
353. 3-CHLORO-N-t8-{ l-[2,3-DlMETHYL-4-(2-MORPHOLlNE-2- YLETHOXYJPHENYLJETHYLj-δ-AZABICYCLOtS^. ηOCT-S-Y^^-FLUOROBENZAMIDE

Step 1. 2-Morpholin-2-yiethanol The previously described 6-(2-hydroxyethyl)morpho!in-3-one (Ll g, 7.57 mmol) is suspended in 50 mL of THF after being crushed to a fine powder. Cooling to 0 0C is followed by slow addition of Red-Al® (65% in toluene, 9.1 mL, 30.3 mmol). The reaction mixture is allowed to warm to rt over night, then cooled back Io 0 0C and quenched with 0.59 mL of water and ] .18 mL of 4N NaOH. After wanning to rt over 30 min, the reaction mixture is stirred for 90 min and then evaporated under reduced pressure. The residue is used in the next step without any further purification.
Step 2. tert-Butyl 2-(2-hydroxyethyl)morpholine-4-carboxylate
The crude mixture from Step 1 (100% = 7.57 mmol) is suspended in 50 mL of DCM and 40 mL of water. Addition of 10 mL of saturated sodium bicarbonate solution adjusts the pH to 12-13. Addition of Boc2O (2 eq.) and stirring over night at rt led to a slightly yellowish heterogeneous mixture that is poured into 300 mL of water. Extraction with DCM (3 x 100 mL), drying over magnesium sulfate and purification on silica gel with straight EtOAc yields the title compound. LCMS (Method 3) m/z (M+Na):253.98.
Step 3. 2-Morpholin-2-yIethyl 4-methyibenzenesulfonate The purified alcohol from Step 2 (1.55 g, 6.7 mmol) is dissolved in 20 mL of DCE and subsequently treated with TEA (8.41 mL, 60 mmol) and DMAP (82 mg, 0.67 mmol). After cooling to 0 0C, tosyl chloride (1.41 g, 7.37 mmol) is added and the mixture is allowed to warm to rt over night before being put into 200 ml of water and 20 mL of saturated potassium carbonate. Extraction with DCM (3 x 100 mL), drying over magnesium sulfate and final purification on silica gel with EtOAc/DCM/hexane (1/1/2) yields the title compound. LCMS (Method 3) m/z (M+Na): 407.95.
Step 4. tert-Bυtyl 2-{2-[4-(l-{3-[(3-chloro-4-fluorobenzoyl)amino]-8-azabicyclo[3.2.1]oct-8- yl } ethyI)-2,3 -dimethylphenoxyjethyl } morpholine-4-carboxyIate
The purified tosylate from Step 3 (132 mg, 348 μmol), previously described phenol (100 mg, 232 μmol), and tetra-butylammonium iodide (86 mg. 232 μmol) are dissolved in 5 mL of DMF. After addition of NaH (60% in min. oil, 37 mg. 928 μmol), the reaction mixture is stirred at it for 16 h before being put into 150 mL of Et2O and EtOAc (1 :1). The organic layer is washed with water/saturated potassium carbonate (100 mL + 10 mL), water (2 x 100 mL), and then dried over magnesium sulfate. After removal of the solvent, the crude material is subjected to a catch-release chromatography (2.0 g SiOa based benzenesulfonic acid resin. 0,7-0.9 mrnoi/g loading). The compound is put onto the column with EtOAc (3 x 2 mL), then washed with EtOAc (4 mL) and ElOAc/MeOH (20/1. 10 mL) before elution with EtOAc/MEOH/TEA (10/1/1, 20 mL). Final purification on silica gel with DCM/MeOH(2N NH3) (15/1 ) yields the title compound. LCMS (Method 3) m/∑ (M+H):644.26.
Step S. 3-Chloro-N-(8-{ l-[2.3-dimethyl-4-(2-morphoIin-2-ylethoxy)phenyl]ethyl}-8- azabicycio[3.2.1 ]oct-3-yI)-4-fluorobenzamide
The Boc-protected compound from Step 4 (100 mg, 155 μmol) is dissolved in 5 mL of DCM and 2 mL of MeOH. Addition of 4N HCL/dioxane (5 mL) and stirring at rt for 2 h results in complete Boc removal as validated by LCMS. Subsequently, the reaction mixture is put into 100 mL of water, basified with saturated potassium carbonate (10 mL), extracted with DCM (3 x 100 mL), and dried over magnesium sulfate. Final purification on silica gei with DCM/MeOH (2N NH1) (10/1) yields the title compound. LCMS (Method 3) m/∑ (M+H):543.27.
354, 3-CHLORO-N~{8-[l-(2,3-DIHYDRO-l-BENZOFURANE-5-YL)ETHYL]-8- AZAB ICYCLO [3.2.1 ] OCT-3 -YL) -4-FLUOROBENZAMIDE tropane formation


Commercially available 5-acetyi-2,3-dihydrobenzofuran (4.8 g, 29.6 mmol), formic acid
(1.12 mL, 29.6 mmol), and formamide (10.7 mL, 237 mmol) are heated to 180 0C for 6 h or until
LCMS shows complete reaction. The cooled reaction mixture is then put into 300 mL of water and extracted with DCM (3 x 200 mL). Drying over magnesium sulfate and final purification on silica gel with EtOAc/MeOH (20/1) affords the title compound. LCMS (Method 3) m/z (M+H):192.12.
Step 2. l-(2,3-Dihydro-l-benzofuran-5-yl)ethanamine
The purified formamide from Step 1 (4.5 g, 23.5 mmol) is dissolved in 200 mL of MeOH and hydrolyzed to the amine with 55 mL of cone. HCl for 40 h. After evaporating most of the organic solvent, the residual aqueous solution is put into 500 mL of water (pH=l). Non-basic side products are extracted with ethyl ether (2 x 300 mL) and discarded. Adjustment of the pH to 12-13 with 1 ON NaOH is followed by extraction with DCM (3 x 200 mL) and drying over magnesium sulfate. The crude mixture is taken on directly to the next step. LCMS (Method 3) m/z (M- NH2): 147.17. Step 3. δ-f l-^^-Dihydro-l-benzofuran-S-y^ethylJ-S-azabicycloP^.ljoctan-S-one
The conversion of the free amine to the tropanone using in situ generated succinaldehyde and 1,3-acetonedicarboxylic acid follows the procedure outlined earlier in the general tropane synthesis.
Step 4. S-f l^^-Dihydro-l-benzofuran-S-yOethylj-S-azabicycloP^.ljoctan-S-one oxime The conversion of the tropanone to the oxime using 1.1 equivalents of hydroxylamine hydrochloride in pyri dine/water (2/1) follows the procedure outlined in the general tropane synthesis.
Step 5. S-fl^^-Dihydro-l-benzofuran-S-yOethy^-δ-azabicycloP^. lJoctano-amine
The crude oxime (14.7 mmol) is dissolved in 100 mL of w-propanol and evaporated under reduced pressure to remove any residual pyridine. After redissolving in 100 mL of w-propanol, the solution is heated to 70 0C and then treated with sodium (3.38 g, 147 mmol). Upon dissolution (15 min), more sodium (3.38 g. 147 mmol) is added and the heterogeneous mixture is stirred at 70 0C until all metal has disappeared (1 h). Subsequently, the mixture is cooled to rt put into 500 mL of saturated sodium bicarbonate solution, extracted with DCM (3x300 mL), and dried over magnesium sulfate. LCMS control shows conversion of 80-90% in an otherwise clean reaction. To drive the reaction to completion, the crude mixture is re-subjected to sodium/n-propanol reduction at 70 0C (2 x 3.38 g Na). A second work-up under the above conditions yields the title compound with only trace starting material left. No further purification is conducted. LCMS (Method 3) m/z (M+H): 273.08.
Step 6. 3-Chloro-N-{8-[l-(2,3'dihydro-l-benzofuran-5-yl)ethyl]-8-azabicyclo[3.2.1]oct-3-yi}-4- fluorobenzamide
The crude amine from Step 5 (100 mg, 367 μmol) is dissolved in 10 mL of DCM, treated with TEA (150 μL, 1.1 mmol), and cooled to 0 0C. Addition of 3-chloro-4-fluorobenzoyl chloride (85 mg, 440 μmoi) is followed by warming to rt over 1 h and additional stirring for 2 h. After LCJMS shows complete conversion, the reaction mixture is put into 1 10 mL of water/saturated potassium carbonate (10/1), extracted with DCM (3 x 100 mL), and dried over magnesium sulfate. Purification on silica gel with DCM/MeOH(2N NH3) (15/1) yields the title compound. LCMS (Method 3) m/z (M+H): 429.05.
355. 3-CYANO-[l-(2,3-DIHYDRO-BENZOFURANE-5-YL)ETHYL]-8- AZAB1CYCLO[3.2.1 ]OCT-3-YL}BENZAMIDE

This Example illustrates a general procedure for HBTU coupling reactions. The previously described 8-[l ~(2,3-dihydro-l -benzofuran-5-yl)ethyl]-8- azabicycio[3.2.1]octan-3-amine (150 μL of a 0.2N solution in toluene, 30 μmol), 3 -cyano benzoic acid (1 80 μL of a 0.2N solution in DMA. 36 μmol), and TEA ( 90 μL of a 1.0N solution in toluene, 90 μmol) are shaken for 5 min at rt before adding HBTU (225 μL of a 0.2N solution in DMA, 45 μmol). Subsequent shaking for 1 h at rt leads to complete conversion to the amide as determined by LCMS, 0.1 mL of water is added and the homogeneous mixture is evaporated at 50 0C using either IR-Dancer or Genevac. The residue is dissolved in 1 mL of EtOAc before 0.5 mL of a water/saturated potassium carbonate-mixture (4/1) is added. The organic layer is decanted and put onto a 0.5 g pre-packed column of silica based benzenesulfonic acid resin (loading -0.9 mmol/g). The remaining aqueous layer is extracted with two more 3 mL portions of EtOAc, which are also deposited onto the column. The loaded column is washed with EtOAc (2 mL) and 5 mL of EtOAc/MeOH (20/1) before the product is eluted with 10 mL of EtOAc/MeOH/TEA and concentrated under reduced pressure. To remove any residual TEA, the dried sample is redissolved in 10 mL of DCM and stripped of its solvent twice. Final purification on silica gel with DCM/MeOH(2N NH3) (15/1) leads to the title compound. LCMS (Method 3) m/z (M+H):402.06.
356. N-(S-[I -(2,3-DIHYDRO- I -BENZOFURANE-S-YL)ETHYL]-S-AZABICYCLOP ^ . I]GCT- 3-YL}-6-METHYLPYRIDlNE-2-CARBOXAMIDE

The procedure follows the genera! reaction for HBTU-coυplings outlined above. The final purification is conducted on silica gel with DCM/MeOH(2N NH3) (15/1). LCMS (Method 3) m/z (M+H): 392.07.
357. 3-CYANO-N-(S- { H2,3-DIMETHYL-4-(MORPHOLINE-2- YLMETHOXY)PHENYL]ETHYL) -S-AZABICYCLOP^- I ]OCT-S -YL)BENZAMIDE

Py/water

Step 1. S-tl^-Hydroxy^J-dimethylphenyOethyiJ-δ-azabicyciop^. ^octan-S-one
The previously described 8-[l-(4-methoxy-2,3-dimethyiphenyl)ethyl]-8- azabicyclo[3.2. I]octan-3-one (6.8 g, 23.7 mmol) is dissolved in 50 mL of DCM and treated with a l .ON HCl/ethyl ether solution (48 mL, 48 mmol). The heterogeneous mixture is stirred for another 10 min and then evaporated. The resulting solid is suspended in 50 mL of DCM and cooled to -78 0C before adding neat boron tribromide (8.95 mL, 94.6 mmol). After warming to rt over 1 h, the reaction mixture is stirred for another 20 h or until judged complete by LCMS. Subsequently, the brownish liquid is put onto 100 mL of ice and 200 mL of saturated sodium bicarbonate solution. Filtering through 10 g of Celite®, extraction with DCM (3 x 200 mL), and drying over magnesium sulfate yields a crude material that is purified on silica gel with EtOAc/TEA (5%) to afford the title compound. LCMS (Method 3) m/z (M+H): 274.09.
Step 2. tert-Butyl 2-({2.3-dimethyl-4-[l-(3-oxo-8-azabicyclo[3.2.1]oct-8-yl)ethyl]phenoxy} methyl)morpholine-4-carboxylate
The purified phenol from Step 1 (273 mg, 1 .0 nimol), previously described tosylate (371 mg, 1.0 mmol), and te tr a~buty\ammonium iodide (369 mg, 1.0 mmol) are dissolved in 3 mL of DMF, then treated with cesium carbonate (0.98 g, 3.0 mmol) and heated to 85 0C for 2.5 h. The resulting suspension is put onto 150 mL of EtO Ac/ethyl ether (1/1), extracted with water/saturated potassium carbonate (10/1, 110 mL) and water (2 x 100 mL). Drying over magnesium sulfate and purification on silica gel with EtOAc/hexane/TEA (1/1/4%) yields the title compound. LCMS (Method 3) m/z (M+H):473.24. Step 3. terr-Butyl 2-[(4-{ l-[3-(hydroxyiinino)-8-azabicyclo[3.2.1]oct-8-yl]ethyl}-233-dimethyl- phenoxy)methyi]morpholine-4-carboxylate
Fonnation of the oxime with hydroxyiamine hydrochloride in pyridine/water (2/1 ) follows the standard protocol as described earlier. No further purification of the crude material is undertaken. LCMS (Method 3) m/z (M+H):488.26. Step 4. tør/-Butyl 2-({4-[l -(3-amino-8-azabicyclo[3.2.1Joct-8-yl)ethy]]-2,3-dimethylphenoxy} m ethy I )m orphol iπe-4-car boxy 1 ate
The crude oxime from Step 3 (100% = 780 μmol) is dissolved in 50 mL of M-PrOH and then evaporated to dryness to remove residual pyridine. This material is dissolved in 100 mL of «-PrOH and heated to 70 0C. A large excess of sodium (>50 eq.) is added and the homogeneous mixture is stirred at 70 0C for 15 min or until all solids disappear. After adding another quantity of sodium (>50 eq.) the heating is continued for an additional Ih or until all metal dissolves. Subsequently, the clear solution is cooled to rt, put into 500 mL of saturated sodium bicarbonate, and then extracted with DCM (3 x 200 mL). The conversion of the reduction is assessed by LCMS and the sodium/n- PrOH reduction is repeated another 2 times or until about 95% complete. The isolated crude amine is not further purified and taken on directly to the next step. LCMS (Method 3) m/z (M+H): 474.32.
Step 5. tert-Butyl 2-{[4-(l-{3-[(3-cyanobenzoyl)amino]-8-azabicycIo[3.2.1]oct-8-yl}ethyl)-2,3- dimethy Iph en oxy] methy 1 } m orpho 1 i n e-4-carboxy 1 ate
The conversion of the crude amine from Step 4 to the amide follows the previously outlined HBTU-coupling procedure and is carried out on a 36 μmol scale. The reaction product is used directly in the next step. LCMS (Method 3) m/z (M+H):603.34.
Step 6. 3-Cyano-N-(8-{ 1 -[2,3-dimethyl-4-(morpholin-2-ylmethoxy)phenyl]ethy!}-8-azabicycIo [3.2.1 ]oct-3-yi)benzamide
The crude amide from Step 5 (100% = 36 μmol) is dissolved in 5 mL of DCM and 2 mL of
MeOH. Addition of 2 mL of 4N HCl/dioxane yields the amine after 1.5 h at rt. The clear mixture is put into 110 mL water/saturated potassium carbonate (10/1), extracted with DCM (3 x 100 mL),
dried over magnesium sulfate, and purified on silica gel with DCM/MeOH (2N NH3) (10/1) to yield the title compound. LCMS (Method 3) m/z (M+H): 503.26.
358. N-(8- { 1 -[2.3 -DTMETHYL-4-(MORPHOLlNE-2- YLMETHOX Y)PHEN YL] ETHYL} -8 -
AZABlCYCLO[3.2.1]OCT-3-YL)-5-ETHYLNICOTINAMIDE

The previously described
 2-({4-[l-(3-amino-8-azabicyclo[3.2.1]oct-8-yl)ethyl]- 2,3-dimethyiphenoxy}methyi)morphoIine-4-carboxylate is used in the standard HBTU protocol (3 6 μmol scale) to yield the amide which is converted under the above conditions to the free secondary amine. The final purification uses silica gei with DCM/MeOH(2N NH3) (10/1) to yield the title compound. LCMS (Method 3) m/z (M+H): 506.33.
2-({4-[l-(3-amino-8-azabicyclo[3.2.1]oct-8-yl)ethyl]- 2,3-dimethyiphenoxy}methyi)morphoIine-4-carboxylate is used in the standard HBTU protocol (3 6 μmol scale) to yield the amide which is converted under the above conditions to the free secondary amine. The final purification uses silica gei with DCM/MeOH(2N NH3) (10/1) to yield the title compound. LCMS (Method 3) m/z (M+H): 506.33.
359. N-(8-{ l-r2,3-DIMETHYL-4-(MORPHOLIN-2-YLMETHOXY)PHENYL]£THYL}-8-
AZABICYCLO[3.2.1 ]OCT-3-YL)-6-METHYLPYRlDINE-2-CARBOXAMIDE
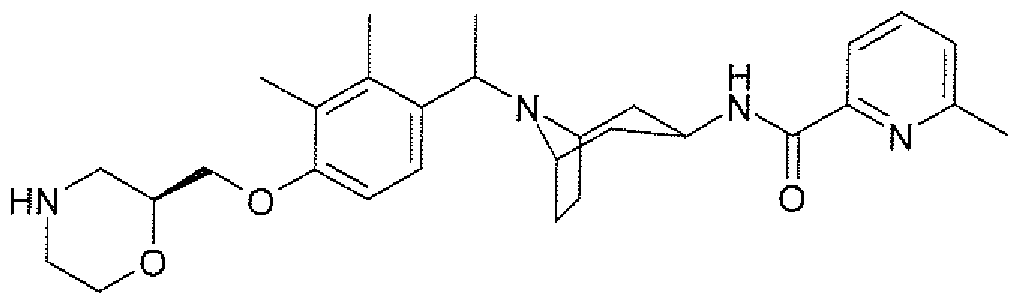
The previously described tert-huty\ 2-({4-[l-(3-amino-8-azabicyclo[3.2.1]oct-8-yl)ethyl]- 2,3-diraethylphenoxy}methyl)morρholine-4-carboxylate is used in the standard HBTU protocol (36 μmol scale) to yield the amide, which is converted under the above conditions to the free secondary amine. The final purification uses silica gel with DCM/MeOH(2N NH3) (10/1) to yield the title compound. LCMS (Method 3) m/z (M+H): 493.37.
360. N-(8-{ 1 -[2,3-DlMETHYL-4-(MORPHOLIN-2-YLMETHOXY)PHENYL]ETHYL}-8-
AZAB1CYCLO[3.2.1 ]OCT-J-YL)-S-FLUORO-O-METHYLPYRIDrNE^-CARBOXAMIDE

The previously described tør/-butyl 2-({4-[l-(3-amino-8-azabicyclo[3.2.I]oct-8-yl)ethyl]- 2,3-dimethyiphenoxy}methyI)morρholine-4-carboxyIate is used in the standard HBTU protocol (36 μmol scale) to yield the amide which is converted under the above conditions to the free secondary
amine. The final purification uses silica gel with DCM/MeOH(2N NH3) (10/1 ) to yield the title compound. LCMS (Method 3) m/z (M+H): 51 1.28.
361. 3-CYANO-N-[S-(I -{4-[4-(DIMEraYLAMlNO)BUTOXY]-2,3-DIMETHYLPHENYL}EΪHYL)-
AZABICYCLO[3.2.1] OCT-3 -YL]BENZAMIDE

Step 1. N-(8-{ l-[4-(4-Chlorobutoxy)-2,3-dimethyIphenyl]elhyl}-8-azabicyclo[3.2.1]oct-3-yI>232,2- trifluoroacetamide
The previously described phenol (2.0 g, 5.4 mmoi) and cesium carbonate (2.64 g, 8.1 mmol) arc suspended in 20 mL of DMF. Addition of 1 -bromo-4-chlorobutane is followed by stirring overnight or until the reaction is judged complete by LCMS. The heterogeneous mixture is put into 200 rnL of EtOAc/ethyl ether (1 : 1) and washed with water (3 x 200 mL). Drying of the organic phase over magnesium sulfate and evaporation of the solvent yields a crude material that is carried on directly to the next step. LCMS (Method 3) m/z (M+H): 461.34.
Step 2. N-[8-(1 -{4-[4-(Dimetliylamino)butoxy]-2,3-dimethylpheny]}ethyI)-8-azabicyclo[3.2.1] oct-3-yi]-2.2,2-trifluoroacetamide
The crude chloride from Step 1 (1.0 g, 2.17 mmol), is dissolved in an excess of a dimethylamine/acetonitrile solution. Heating to 80 0C overnight results in clean conversion to the tertiary amine. The light yellow mixture is put into 300 mL of water, extracted with DCM (2 x 200 mL), and dried over magnesium sulfate. Evaporation of the solvent yields a crude material that is carried on directly to the next step. LCMS (Method 3) m/z (M+H): 470.30.
Step 3. S^l -^^-^DimethylaminoJbutoxyJ^^-dimethylphenyllethyO-S-azabicycloP^.l] octan-3 -amine
The crude tertiary amine from Step 2 is dissolved in 30 mL of MeOH and then treated with
ION NaOH (0.95 mL, 9.5 mmol). Stirring overnight leads to full conversion to the free primary amine as judged by LCMS. After evaporation of the solvent under reduced pressure, the residue is put into 120 mL of brine/saturated potassium carbonate (5/1) and extracted with DCM (3 x 100 mL).
The combined organic layers are dried over magnesium sulfate and evaporated to yield a crude materiel that is used directly for subsequent amide formations without any further purification. LCMS (Method 3) m/z (M+H): 374.28.
Step 4. 3-Cyano-N-[8-(l-{4-[4-(dimethylamino)butoxy]-2,3-dimethylphenyl}ethyl)-8-azabicyclo [3.2. I]oct-3-yl]benzamide
The primary amine from Step 3 (150 μL of a 0.2N solution in toluene, 30 μmoi), 3- cyanobenzoic acid (180 μL of a 0.2N solution in DMA, 36 μmol), and TEA (90 μL of a 1.0N solution in toluene, 90 μmol) are shaken for 5 min at rt before adding HBTU (225 μL of a 0.2N solution in DMA, 45 μmol). Subsequent shaking for 1 h at it leads to complete conversion to the amide as checked by LCMS. 0.1 mL of water is added and the homogeneous mixture is evaporated at 50 0C using either IR-Dancer or Genevac. The residue is dissolved in 1 mL of EtOAc before 0.5 mL of a water/saturated potassium carbonate-mixture (4/1) is added. The organic layer is decanted and put onto a 0.5 g pre-packed column of silica based benzene sulfonic acid resin (loading -0.9 mmol/g). The remaining aqueous layer is extracted with 2 more 1 mL portions of EtOAc which are also deposited onto the column. The loaded column is washed with EtOAc (2 mL) and 5 mL of EtOAc/MeOH (10/1 ) before the product is eluted with 10 mL of EtOAc/MeOH/TEA and concentrated under reduced pressure. To remove any residual TEA, the dried sample is dissolved in 10 mL of DCM and stripped of its solvent twice. Final purification on silica gel with EtOAc/MeOH/TEA (20/1/1 ) leads to the title compound. LCMS (Method 3) m/z (M+H): 503.37.
362. AMΪDΛΉON OF 8-(l -{4-[4-(DlMETHYLAMINO)BUTOXY]-2,3-DIMETMYLPHENYL} ETHYL)-S- AZABICYCLO[3.2.1 ]OCTAN-3 -AMINE AND RELATED COMPOUNDS
Amidation reactions resulting in the compounds in Table Il are carried out using the procedure described in the 4Ih step of the previous example, with readily apparent modification of stalling material. In most cases, the purity of the listed compounds does not warrant any purification other than the basic work-up. Hence, the silica gel step is not performed but the compounds are tested as crude materials. In Table 11, compounds with a "*" in the column headed "K5" have a Kj in the assay of Example 10 that is less than 1 micromolar. The determined molecular weight (presented as M+L determined using Method 3) is shown in the column headed "MS" in Table II, along with the retention time in minutes.
Table Il
Compound Name K1 i T_R MS

* 1.09 508.28 oct-
 methoxybenzamide ]
methoxybenzamide ]
365 * 1.13 510.27

ino) butoxy ] ethyl)- I . I l 514.25 .1 ]oct-
 difiuorobenzamide ]
difiuorobenzamide ]
1.1 1 522.28
 ethoxybenzamide
ethoxybenzamide


3-yl]benzamide
Compound Name K1 _ TLR: MS
1.10 538.28
1.12 542.22


(trifϊuoromethyl)benzam ide ]
1.1 1 496.30
 furamide ]
furamide ]
375 I)- 1 .08 482.28



* 1 .15 526.25


1.07 484.23
 ]
]
1.07 484.24 oct-

1 )- 1.13 543.22
 methoxyisonicotinamide
methoxyisonicotinamide

I)-
1.09 470.32
 e
e
386 1.11 484.33
 e
e

3 ethyl)- 1.12 5 Ϊ0.27 .1 ]oct-
 methyibenzamide
methyibenzamide

(methylthio)thiophene-
2-carboxamide ] I)- 1.07 493.29
id ine-
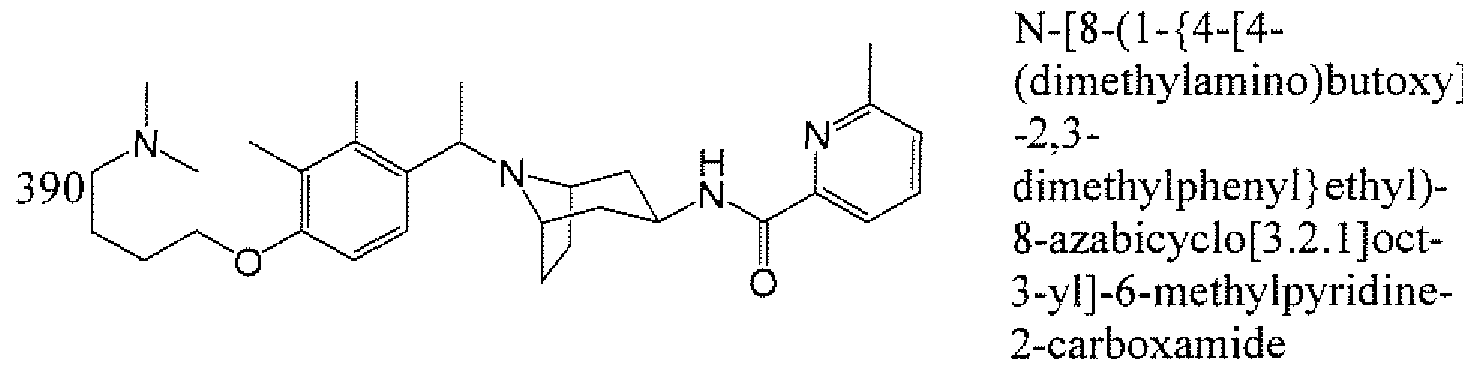 -{4-[4- butoxy] ethyi)- 1.10 557.15 . l]oct-
-{4-[4- butoxy] ethyi)- 1.10 557.15 . l]oct-

 ] I)- * 1.13 506.30
] I)- * 1.13 506.30
 d i methy lbenzam i de ]
d i methy lbenzam i de ]
1.13 506.30
 dimethylbenzamide
dimethylbenzamide
95 I)- 1.10 547.24


(trifluoromethy])pyrimid ine-5-carboxamide
1.12 547.24

(trifluoromethyl)pyridin e-2-carboxamide butoxy] ethyl)- 1.1 1 492.30 .1 ]oct-
 methyibenzamide
methyibenzamide

)- 1.15 536.29 oct-
 phenoxy )acetam ide
phenoxy )acetam ide
I)- * 1.03 493.29
 methylnicotinamide
methylnicotinamide
402 1.03 469.26
 carboxamide ]
carboxamide ]
.06 497.28 oct-
 oxazole-5-carboxamide
oxazole-5-carboxamide
EXAMPLE 4. PREPARATION OF ADDITIONAL REPRESENTATIVE AMINOPIPERIDINES AND RELATED COMPOUNDS
Compounds in Tables III and IV are prepared using the methods described above. In some cases, synthetic modifications and/or additional steps that will be readily apparent are employed. For example, certain compounds are prepared from starting materials such as substituted pyridine amides (e.g., 6-bromopicolinic amide or 5-bromonicotinic amide). Modifications of the above procedures to allow the use of such starting materials will be readily apparent.
In Tables III and IV, compounds with a "*" in the column headed "K1" have a K1 in the assay of Example 10 that is less than 1 micromolar. The determined molecular weight (presented as M+l) is shown in the column headed "MS" in Tables III and IV, along with the retention time in minutes. In Table III, the retention times for compounds analyzed via HPLC MS Method 2 or Method 3
provided above include the method number in parentheses, and the retention times for compounds analyzed via HPLC MS Method 1 provided above are unmarked. In Table IV, MS Method 3 was used for all data provided.
Table IH

2-(1-beπzy!-1 H-tmιdazo!- 4-yl)-N-[8-(4-methoxy-
405 2,3-dtmethyfbenzyl)-8- 1 13 473 08 azabιcyclo[3 2 1]oct-3- yl]acetamιde



2-(2,4-dichiorophenyl)-N- {8-[4-(2-methoxyethoxy)- 12 2,3-dimethylbenzyl]-8- 1.22 505.13 aza bιcyclo[3.2.1 ]oct-3- yljacetamide


Compound Name K, Time MS

2-(2,5-dimethyiphenyi)-N- {8-[4-(2-rnethoxyethoxy)- 2t3-dimethylbenzyl]-8- 1.22 465.25 azabicyclo[3.2.1 ]oct-3- yljacetamide
2-(2,6-dichlorophenyl)-N-
{8-[4-(2-methoxyethoxy)-
2,3-dimethyJbenzy[]-8- 1.2 505.13 azabicyclo[3.2.1]oct-3- yl}acetamide
 Ret
Ret
Compound Name K,- Time MS

2-(2-bromophenyl)-N-{8~
[4-(2-methoxyethoxy)-
421 2,3-dimethylbenzyl]-8- 1.2 515.12 azabicyc!o[3.2.1]oct-3- yl}acetamide



Compound Name K, Time MS
2-(3,4-dιffuorophenyl)-N- {8-[4-{2-methoxyethoxy)-
«9 2,3-dιmethy[benzyl]-8- * 1 19 473 17 azabιcyclo[3 2 1]oct-3- yl}acetamιde


Compound Name K1 Time MS

2-(4-bromophenyl)-N-{8- [4-{2-methoxyethoxy)'
438 2,3-dιmethytbenzyl]-8- 1 22 515 12 azabιcyc[o[3 2 1 joct-3- yl}acetamιde


Compound Name K, Time MS

 3-yl)- 3- 1 07 472 25
3-yl)- 3- 1 07 472 25

2,5-dιfluoro-N-{8-[4-(2- methoxyethoxy)-2,3- dιmethylbenzyl]-8- 1 17 459 19 azabιcyclo[3 2 1]oct-3- yljbenzamide


Compound Name K1 Time MS
 Ret
Ret
Compound Name K, Time MS
456 1 26 557 16
457 1 15 523 24
458 1 24 539 17
459 1 21 523 19
460 1 19 513 19
461 1 17 504 14
 Ret
Ret
Compound Name K, Time MS

3,4-dιchloro-N-{8-j;4-(2- methoxyethoxy)-2 , 3- dιmethylbenzyl]-8- 1 24 491 12 azabιcyc!o[3 2 1]oct-3- yi}beπzamιde
 Ret
Ret
Compound Name K, Time MS
3,4-dιfluoro-N-[8-{3-
468 fluoro-4-methoxybenzyl)- * 1 17 405 21 8-azabιcyclo[
3,4-dιfluorα-N-{8-E4-(2- methoxyethoxy)-2,3-
469 dimethyibenzyi]-8- 1 19 459 16 azabιcyclo[3 2 1]oct-3- yl}benzamtde
3,5-dιmethoxy-N- [(1S,3R)-8-(4-methoxy-
470 2,3-dsmethylbenzyl)-8- 1 13 439 21 azabιcyclo[3 2 ijoct-3- yljbenzamide
3,5-dιmethoxy--N-[8-{4- methoxy-2,3-
471 dimethylbenzyl}-8- 1 16 453 19 azabιcyclo[3 2 1]oct-3-yi]- 4-methylbenzamιde
3,5-dιmethoxy-N-[8-(4- methoxy-2,3-
472 dιmethylbenzyS)-8- 1 2 439 32 azabιcyclo[3 2 1]oct-3- yljbeπzamide
 Ret
Ret


3-chloro-4-fluoro-N-{8-[4- (2-hydroxyethoxy)-2,3- dιmethylbeπzyl]-8- 1 17 461 12 azabicyclo[3 2 1]oct-3- yljbenzamide
 Ret
Ret
Compound Name K= Time MS

3-chloro-N-(8-{4-[3- (dimethylamino}propoxy]- 2,3-dιmethylbenzyl}-8- 1 13 502 32 azabιcyc(o[3 2 1]oct-3- yl)-4-fluorobenzamide
3-chloro-N-(8-{4-[3- (ethylamιno)proρoxy]- 2,3-dιmethylbeπzyl}-8- 1 14 502 36 azabιcyclo[3 2 1]oct-3- yl)-4-f[uorobenzamιde
 Ret
Ret

Compound Name K, Time MS

502 oxy}-2,3-dιmethy!benzyl)- 1 13 516 38 8-azabιcyclo[3 2 1]oct-3- yl]-4-f[uorobenzamιde
3-chloro-N-[8-{4- methoxy-2,3-
503 dιmethy!benzyl)-8- 1 11 413 11 azabιcyclo[3 2 1]oct-3- yl]beπzamϊde
3-chloro-N-{8-[2T3- d[methyl-4-(2-morpho[iπ-
504 4-ylethαxy)beπzyl]-8- 1 03 530 29 azabicyclo[3 2 1]oct-3- yl}-4-fluorobenzamιde


508 17
509 5


3- 13 12 45519 -3- ide
 Ret
Ret

Compound Name K1 Time MS
4-chloro-N-{8-[4-(2- methoxyethoxy)-2 , 3-
520 dιmethyfbenzyl]-8- 1 23 471 17 azabicyclo[3 2 1]oct-3- ylJ-3-methylbenzaπvde
4-chloro-N-{8-[4-(2- methoxyethoxy)-2,3-
521 dιmethylbenzyl]-8- 1 2 457 15 azabicycio[3 2 1]oct-3- yl}benzamide
4-fluoro-N-{8-[4-(2- meth oxyeth oxy )-2 , 3-
522 dimethylbenzyt]-8- 1 21 455 22 azabιcycto[3 2 1]σct-3- yi}-3-methylbenzam[de
4-fluoro-N-{8-[4-(2- m ethoxyet hoxy ) -2 , 3-
523 dιmethylbenzyl]-8- 1 19 486 40 azabιcyclo[3 2 1]oct-3- yl}-3-nitrobenzamide
4-fluoro-N-{8-[4-(2- methoxyethoxy)-2,3-
524 dιmethylbenzyl]-8- 1 17 441 17 azabϊcyclo[3 2 1]oct-3- yljbenzamide
5,6-dιchloro-N-{8-[4-(2- methoxyethoxy)-2,3-
525 dιmethylbenzyl]-8- 1 2 492 15 azabscyclo[3 2 1]oct-3- yljmcotinamide
 Ret
Ret
Compound Name K, Time MS
5-bromo-2-ch!oro-N-{8- [4-(2-methoxyethoxy)-
526 2,3-dimethylbenzyl]-8- 116 53810 azabιcyclo[3 2 1]oct-3- yl}nιcotιπamide
5-bromo-6-chloro-N-{8- [4-(2-methoxyethoxy)-
527 2,3-dimethylbenzy[]-8- 121 53811 azabιcyclo[3 2.1]oct-3- yl}nιcotιπamide


Compound Name K, Time MS

N-[8-(4-methoxy'2,3~ dιmethylbenzyl)-8-
536 azabιcyc(o[3 2 1]oct-3-yl]- 1 27 512 29 2-[(3,5,6-trιchioropyndιn- 2-yl}oxy]acetamιde
N-[8-(4-methoxy-2,3- dimethylbenzyl)-8-
537 azabιcyclo[3 2 1]oct-3-yl]- 1 11 466 01 2-[2-(1 ,3-thιazol-2-yl)-1 H- !midazol-1-yl]acetarri!de
 Ret
Ret
Compound Name Kj Time MS

N-[8-(4-methoxy-2,3- dimethyibenzyl)-8-
539 azabicyclo[3.2.1 ]oct-3-yl]- 1.14 461.05 2-[4-{1 H-tetrazoi-1- yl}phenyl]acetamide


Compound Name K, Time MS

1 22 521 18 l]
* 1 2 581 19 -
 Ret
Ret
Compound Name K= Time MS
549 3- 1 21 505 19 yl]ac

550 1 23 597 18 p he ny
551 1 23 521 18

552 1 22 581 20
3-
553 1 15 521 17

Compound Name K1 Time MS
1.21 505.19 c
1.2 483.19

556 1.23 597.18
557 1.18 551.16
558 1.23 521.18

Compound Name K1 Time MS
 Ret
Ret

Compound Name K, Time MS




Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K, Time MS

 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS
612 116 44312 ιde
613 117 43717
, 3-
614 116 42316

4-methoxy-N-{8-[4-(2- methoxyethoxy)-2,3-
615 dimethylbenzyl]-8- 117 45316 azabιcyclo[321]oct-3- y[}benzamιde
 l)-
l)-
6 υ1i6υ 11648115
617 117 45316
18 114 46416
115 49017 19 115 4901/ -
20 116 44115

 Ret
Ret
Compound Name K, Time MS
2-{2,4-dιfluoropheπyi)-N- (8-[4-(2-methoxyethoxy)-
629 2,3-dimethylbenzyi]-8- 1 18 473 17 azabιcyclo[3 2 1]oct-3-
 yl}acetamide
yl}acetamide

Name K, Time MS
2-(4-ethoxyphenyl}-N-{8- [4-{2- meth oxyethoxy ) -2 , _
637 dιmethy[benzyi]-8- azabicyclo[3.2 1]oct-3-
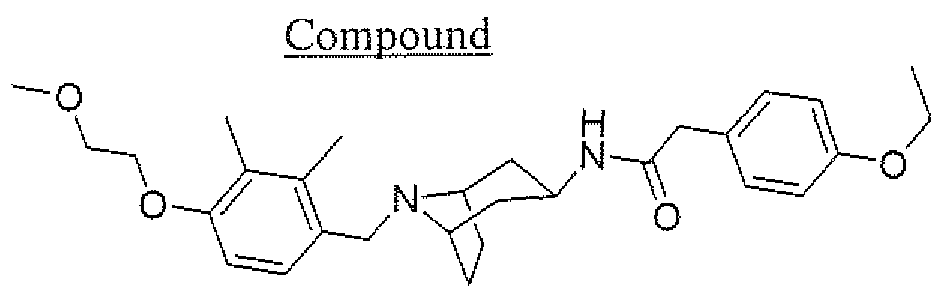 yijacetamide
yijacetamide

4-ethyl-N-{8-[4-(2- methoxyethoxy)-2,3-
640 dimethylbenzyl]-8- 1 21 451 22 azabicyclo[3 2 1]oct-3-
 yljbenzamide
yljbenzamide

Compound Name K, Time MS

651 -y!}- 1 21 505 19 ljace
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS

N-{1-[1-(4-methoxy-2,3- dimethylpheny[)ethyl]pyrr
658 olidin-3-y!}-5- 1.23 422.12
(tπfluoromethyl)pyridine-
2-carboxamide



2-methoxy-N-({1-[1-{4- methoxy-2,3~
664 dimethylpheπyl}ethyl]pipe 1.13 411.33 ridin-3- yl}methyl)benzamide
 Ret
Ret
Compound Name K, Time MS
3,4-dιmethoxy-N-({1-[1-
(d-φethoyy-2,3-
665 d[methylphenyl)ethyj]pipe 11 44136 rιdιn-3- yl}methyl)benzamtde
3,5-dιmethoxy-N-({1-[1-
(4-methoxy-2t3-
666 dιmethylphenyl)ethyl]pipe 11444135 rιdin-3- yl}methy[)benzamfde
213-dιmethoxy-N-({1-[1-
(4-methoxy-2,3-
667 dimethylphenyl)ethy!]p!pe 11244135 rιdιn-3- yl}methyl)benzamtde
3-f!uoro-N-({1-[1-(4- methoxy-2,3-
668 dimethylphenyl)ethy!]pipe 114 39931 rιdιn-3- yl}methyl)benzamιde

Compound Name K, Time MS
4-fluoro-N-({1 -l1 -(4- methoxy-2,3-
669 clιmethy!phenyl)ethyl]pιpe 1 14 399 31 rιdin-3- yl}methyl)benzamιde



Compound Name K1 Time MS
4-(dif!uoromethoxy)-N-
({1-[1-{4-methoxy-2,3-
677 dimethylpheny[)ethyl]pιpe 1 14 447 32 rιdin-3- yl}methyl)benzamicie


1 -[1 -(4-
679 hyl]pιpe 1 17 439 37 )de

Compound Name K, Time MS
-
680 ιpe 1.16 417.31


Ret
Compound Name K1 Time MS

Ret
Compound Name K1 Time MS
N-({1-[1-(4-methoxy-2,3- cl!methylphenyl)ethyl]pipe
686 ridin-3-yl}methyt)-2- 114 449 31
{trifluoromethyl)benzamicl


Ret
Compound Name K, Time MS

3-chloro-N-{{1-[1-(4- methoxy-2,3-
690 dιmethylphenyl)ethyl]pιpe 1 16 415 29 rιdιn-3- yl}methyl)benzamιde
2,6-dιfIuoro-N-({1-[1-(4- methoxy-2,3-
691 dtmethylphenyl)ethyl]pιpe 1 11 417 31 rtdιn-3- yl}methyl)benzamfde

-[1-(4-
692 hyi]pϊpe 1 12 449 27 ιde
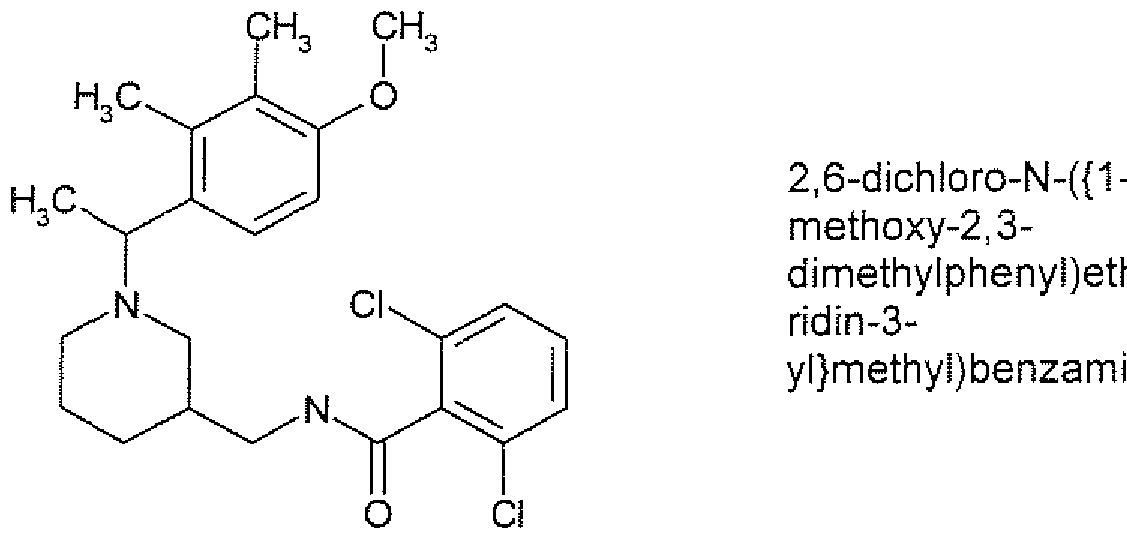 Ret
Ret
Compound Name K1 Time MS
4-chloro-N-({1-[1-(4- methoxy-2,3-
693 dιmethylpheπy!)ethyl]pipe 1 16 415 29 rιdιn-3- yl}methyl)beπzarriϊde

694 1 15 465 31
695 1 19 465 31

Compound Name K1 Time MS
-[1-(4-
696 hy!]pipe 1 18 441 34 !de

697 e 1 21 449 27

(4-
69S pipe 1 16 449 27

N-({1-[1-(4-methoxy-2,3- dιmethylpheπyi}ethyl]pιpe
699 πdιπ-3-yl}methyl)-3- 1 17 449 31 (trιfiuoromethyl)benzamιd
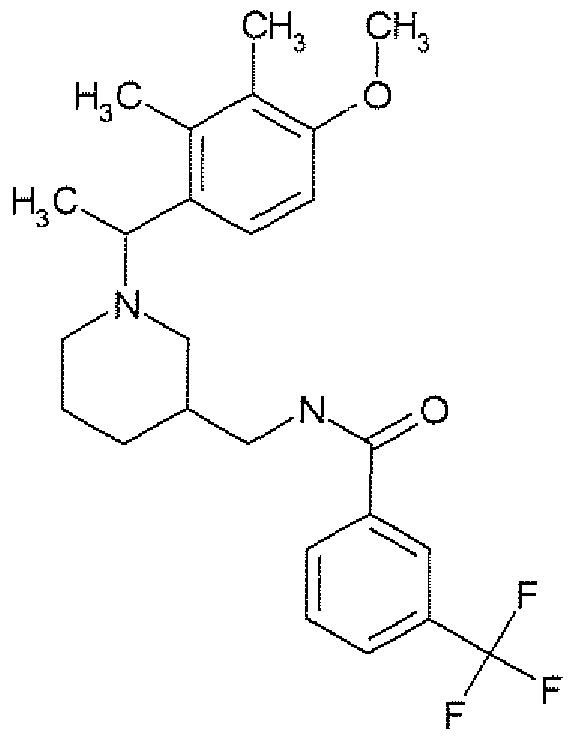
Compound Name Ki Time MS
2,5-dichloro-N-({1-[1-(4- methoxy-2,3- dimethylphenyl)ethyl]pipe 1.15 449.27 ridin-3- yl}methyl)beπzamide

1.18 449.3Ξ

3,4-dichloro-N-{{1-[1-(4- methoxy-2,3- dimethylphenyl)ethyl]pipe 1.2 449.27 ridiπ-3- yl}methyj)benzamide

Compound Name K, Time MS
703 1 17 449 27
704 1 2 423 37

N-({1-[1-(4-methoxy-2,3- dιmethy[pheπyl)ethyl]pιpe
705 rιdιn-3- 1 2 457 37 yl}methyl)bιphenyl-4- carboxamide

Compound Name K, Time MS
113 38219
115 38418
11540216
113 38222

3-chloro-4-fluoro-N-{{1-
[1-(4-methoxy-2,3-
710 dfmethylphenyi)ethyl]pyrr 11941913 olιdm-3- yl}methy[)benzamide
 Ret
Ret
Compound Name K1 Time MS

3-ch!oro-4-f!uoro-N-[1-{1-
{2-methyl-4- i: E(methylamino)carboπyl]p 1 15 432 18 heπyl}ethyl)ptpeπdιπ-4- yljbenzamide


Compound Name K, Time MS (1-{4- mιno)carbo ethyl)pιperι 11745819 de


Compound Name K1 Time MS

3-chloro-4-fluoro-N-{1-[1-
(2-methyl-4-
{[(tetrahydrofuran-2-
723 yimethy!)amino]carbonyl} 1.18 502.20 pheny[)ethyl]piperidin-4- yl}beπzamide


Ret

Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K,- Time MS

 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS

3-chloro-4-f[uoro-N-{1 -[1 -
(2-methyl-4-{[(pyridin-2-
748 ylmethyi)amino]carbonyl} 1.12 509.19 phenyt)ethyl]piperidin-4- yljbenzamide

Compound Name K, Time MS

3-chloro-4-fluoro-N-{1-[1-
(2-methyl-4-{[(pyπdιn-3-
751 yimethyl)amino]carbonyl} 1 11 509 20 phenyl)ethy[]prperιdιn-4- yl}benzamιde
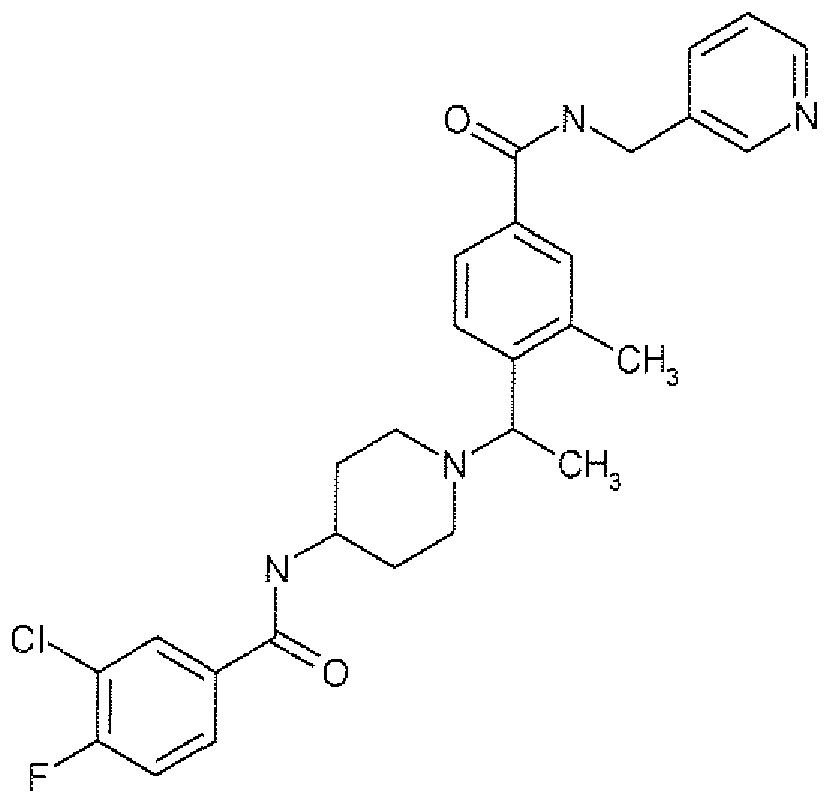
Compound Name K, Time MS
3-chioro-4-fluoro-N-{1-[1-
(2-methyl-4-{[(pyridin-4-
752 yimethyl)amino]carbonyi} 1.1 509.21 phenyl)ethyl]piperidin-4- yl}benzamide


Compound Name K1 Time MS
4-(1-{4-[(3-chloro-4- fluorobenzoyl)amino]-1 -
756 pipeιϊdinyl}ethyl)-N-{2- 1.14 476.18 hydroxyethyi)-N,3- dimethyibenzamide
4-{1-{4-[(3-chioro-4- fluorobenzoyl)amino]-1-
757 piperidinyi}ethyl)-N-(2- 1.17 490.31 methoxyethyl)-N,3- dimethylbeπzamide



764 8

765 117 36906
766 112 35407
767 117 35407
 Ret
Ret
768
769


Compound Name K, Tune MS
 Ret
Ret
Compound Name K1 Time MS

6-fluora-N-{(3R)-1-[1-(4- methoxy-2,3-
777 dimethylpheny!)ethyl]pyrr 1 19 372 05 oiιdιn-3-yl}pyπdme-2- carboxamide
Chiral
3,5-dιfluoro-N-{(3R)-1-[1-
(4-methoxy-2,3-
778 dιmethylphenyl)ethyl]pyrr 1 17 390 03 olιdιn-3-yl}pyrιdine-2- carboxamide
irai
4-chloro-N-{(3R)-1-[1-(4- methoxy-2,3-
779 dimethylphenyljethyljpyrr 1 22 388 01 ohdιn-3-yl}pyπdιne-2- carboxamide
 Ret
Ret

2-fluoro-N-{(3R)-1-[1-(4- methoxy-2,3-
783 dιmethylphenyl)ethyf]pyrr 116 37205 olιdιn-3-yl}nιcotιnaιτi!de
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name Ki Time MS
 Eet
Eet
Compound Name K1 Time MS

Chiral
2-hydroxy-N-{(3R)-1-[1-
(4-methoxy-2,3-
795 dιmethyiphenyl)ethy!]pyrr 126 38308 olιdιn-3-yl}-3- methy!benzamιde

796
797


Chirai
2-hydroxy-N-{(3R)-1-[1- (4-methoxy-2,3-
799 dιrnethy[phenyl)ethy!]pyrr 1 27 434 05 olιdιn-3-yl}-5-( 1 H-pyrrol- 1-y!)benzamιde

Compound Name K1 Time MS
* 1.2 436.31

2-(2-chloro-4- fluorophenyi)-N-{1 -[1 -(4- methoxy-2,3- 1.23433.29 dimethylphenyl)ethyl]-3- piperidinyl}acetamtde
3-ch loro-4-fl uoro-N-{ 1 -[ 1 - (4-methoxy-2,3- dimethylphenyl)ethyl]-3- 1.16419.27 pipeπdinyϊ}beπzamide
3-chloro-4~fluoro-N-{1 - [{1 R)-1-{4-methoxy-2,3- dirnethylpheny!)ethy!]-3- 1.18419.27 piperidinyl}beπzamide
3,5-dimethoxy-N~{(3R)-1- [(1 R)-1-(4-methoxy-2,3- dimethyϊphenyl)ethyl]-3- 1.21 427.24 piperidiny[}benzamide

 Ret
Ret
Compound Name K1 Time MS
1 19 457 31
12645332
11 48434
112 49837
11 47038
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K, Time MS
 Ret
Ret
Compound Name K, Time MS
 Ret
Ret
Compound Name K1 Time MS

833 1.15 528.07
 Ret
Ret
Compound Name K, Time MS

 Ret
Ret
Compound Name K, Time MS
108 38414
11840718
11744322
12 45722
11648221
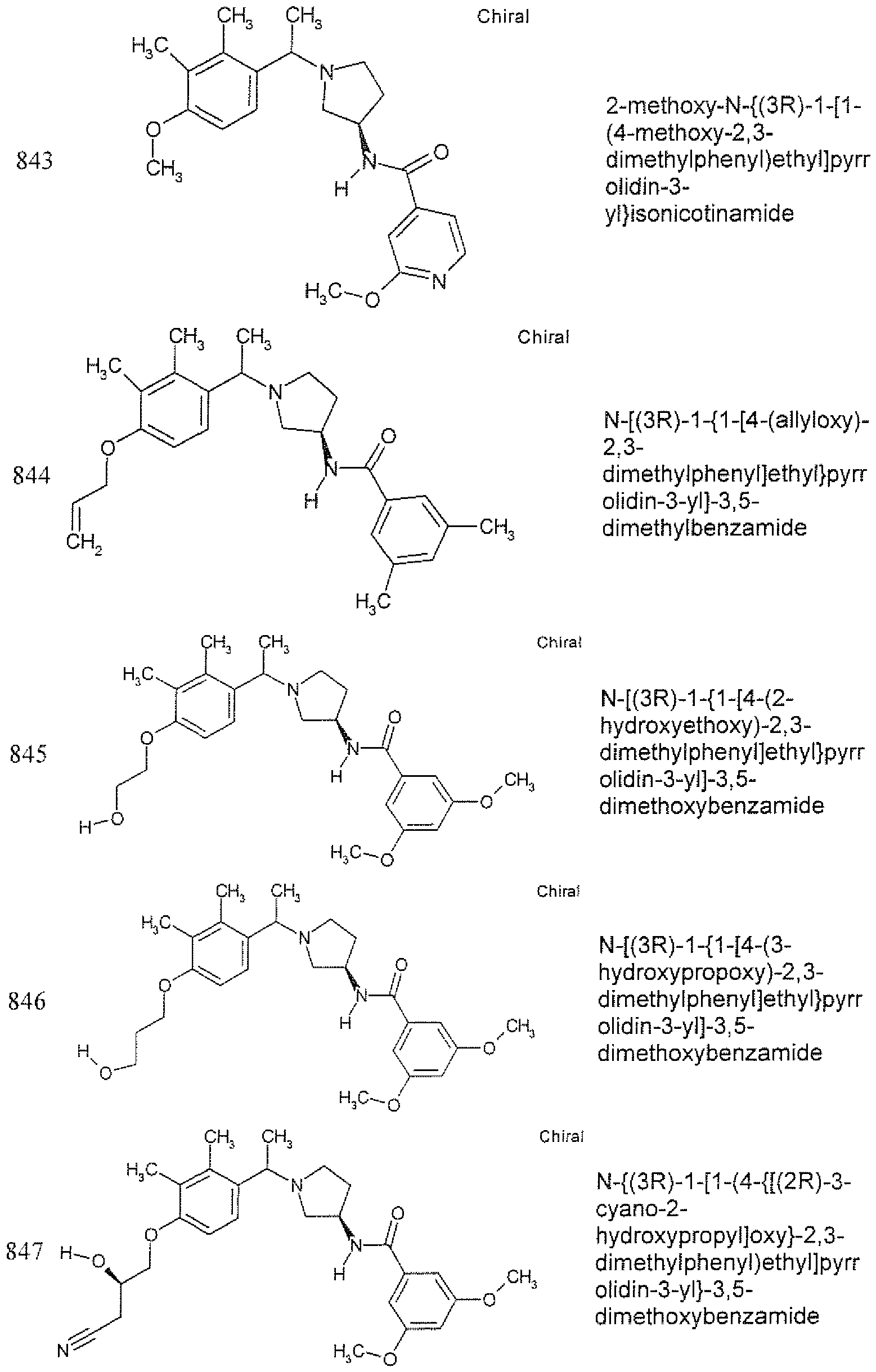 Eet
Eet
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name Kj Time MS
 Ret
Ret
Compound Name K, Time MS

1 05 394 22
1 05 382 22
 Ret
Ret
Compound Name K, Time MS
3-fluoro-N-{(3R)-1-[1-{4- methoxy-2,3-
868 c!ιmethylphenyl)ethyl]pyrr 118 38520 ohdιn-3-yl}-5- methylbenzamide
 ilyloxy)-
ilyloxy)-
869 hy[}pyrr 11 40820 amide


Compound Name K, Time MS

875 n- 118 44335

876 idin- 115 42934


Compound Name K, Time MS
 Ret
Ret
Compound Name K1 Time MS
-
1.13 391.32
1.12 391.33
1.15 405.34
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS

3- ιpe 1 08 395 28

Compound Name K, Time MS
 Ret
Ret
Compound Name K1 Time MS

902 405 32
 Ret
Ret
Compound Name K1 Time MS
903 1.09 377.32
904 1.11 391.34

905 405.36
 Ret
Ret
Compound Name K1- Time MS

-
907 3- 1.17 475.29 ipe

ipe
908 1.14 469.33

Name K1 Time MS
1-acetyl-N-(1-{1-E4-(2- me{hoxyethoxy)-2 , 3-
909 dimethylpheny!]ethy[}pιpe 1 07 460 38 ridιn-4-y[)pipeπdine-4- carboxamide

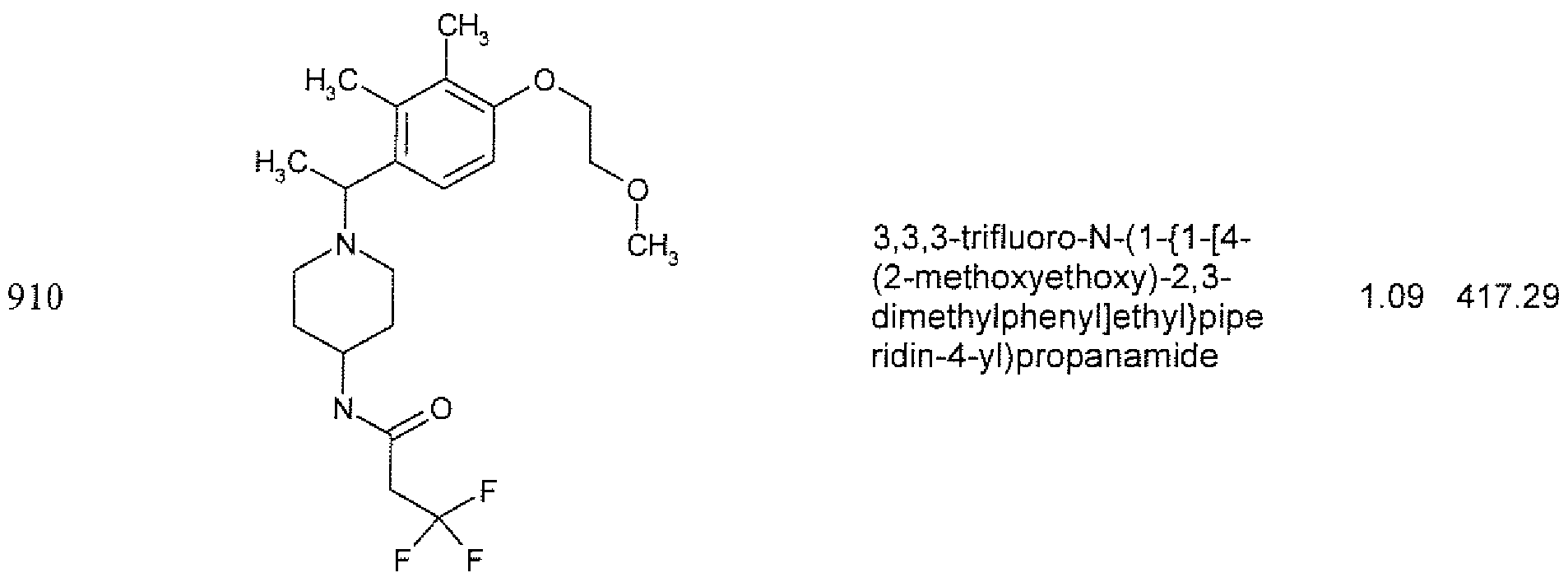
3-
91 ! yi}pfpe 1 13 425 33

Compound Name Ki Time MS
»12 * 1.15 439.35
913 1.14 439.35
914 1.14 439.35


Compound Name K, Time MS
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K, Time MS
 Ret
Ret
Compound Name K, Time MS

112
 Ret
Ret

Compound Name Ki Time MS
 Ret
Ret
Compound Name K, Time MS
 Ret
Ret
Compound Name K,- Time MS
 Ret
Ret
Compound Name K, Time MS
 Ret
Ret
Compound Name K, Time MS

960 449.15
 Ret
Ret
Compound Name K1 lime MS
 Ret
Ret
Compound Name K1 Time MS

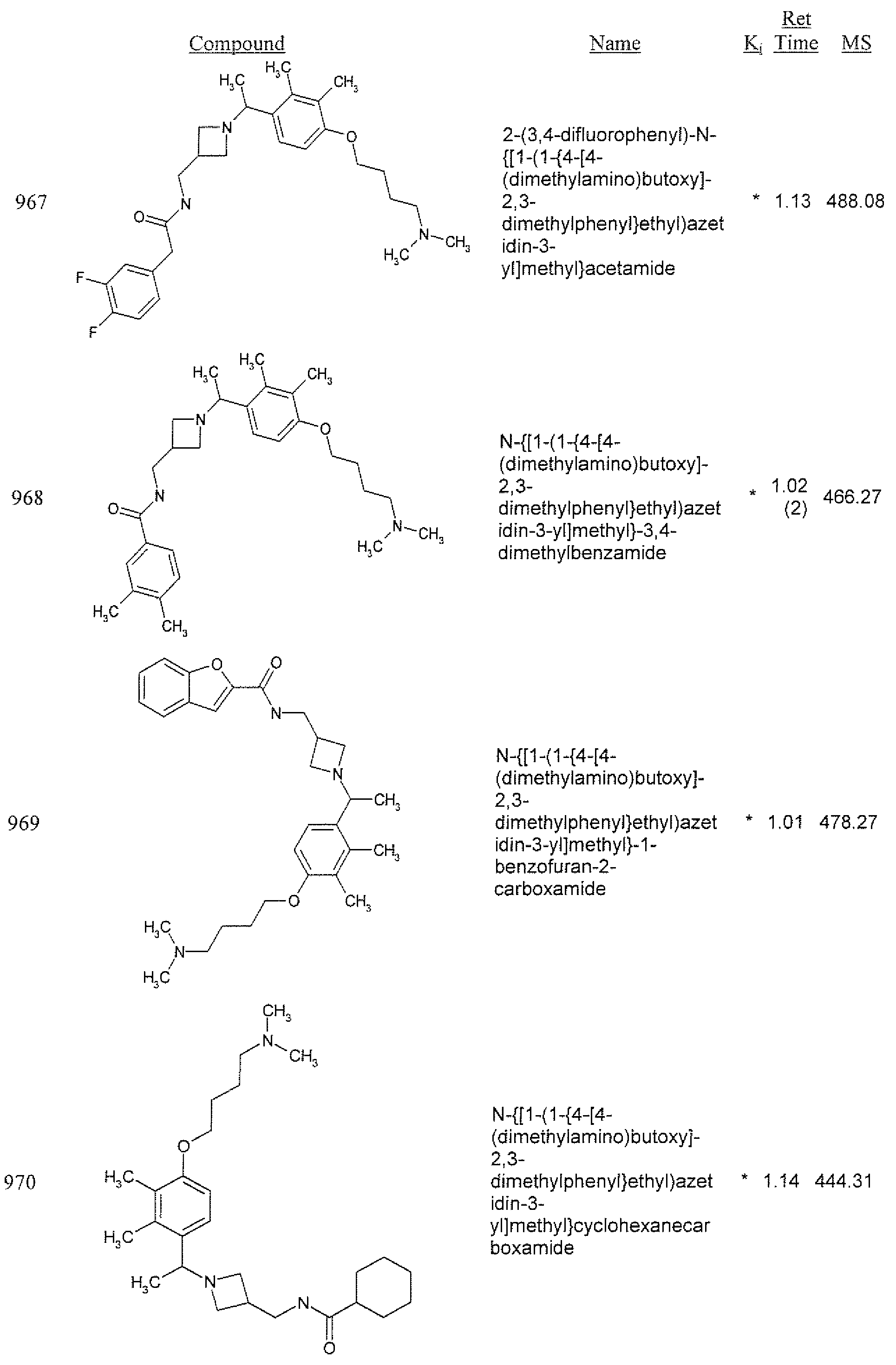 Ret
Ret
Compound Name K,- Time MS
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K, Time MS

N-({1-[1-{4-{[(2R)-2- hydroxypropyi]oxy}-2,3- 81 djmethy[pheny])ethyl]azet * 1 15 411 22 ιdιn-3-yi}methyl)-3- methylbenzamtde

Compound Name Ki Time MS
 Ret
Ret
Compound Name Ki Time MS
 Ret
Ret
Compound Name K1 Time MS
989 et * 1.18 445.18


503.22 479.19
 Ret
Ret
Compound Name K1 Time MS
993 457.22
994 445.17


Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS

N-({1-[1-(4-{[(2R)-2- hydroxypropyl]oxy}-2,3-
1004 dιmethyiphenyl)ethyl]azet 1 11 427 23 ιdϊn-3-yl}methyl)-3- methoxybeπzamide
 Ret
Ret
Compound Name K, Time MS
 Ret
Ret

Compound Name K, Time MS
 Ret
Ret

Compound Name K1 Time MS

Ret
Compound Name K, Time MS
1.14 (2) 41521
113 49609
* 103 49129

Compound Name K1 Time MS

N-({1-[1-(4-{[(2R)-2- hyd roxy pro py l]oxy}-2 , 3-
1028 dιmethyiphenyl}ethy[]azet 1 16 415 21 ιdιn-3-yl}methyl)-2,5- dsmethyl-3-furamide

Ret
Compound Name K, Time MS

Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS

1038 432 18
 Ret
Ret
Compound Name K, Time MS
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K, Time MS
1

N-{{1-[1-(4-{[(2R)-2- hydroxypropy!]oxy}-2,3-
1049 dιmethy!pheπyl)ethyl]azet 114 42721 ιdιn-3-yl}methyl)-3- methoxybenzamide



1054 1 07 523 27


Compound Name K1 Time MS

1058 116 44122
1059 098 48928
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS
1071 431.28


Compound Name K1 Time MS
Ret
Compound Name K1 Time MS

Ret
Compound Name Ki Time MS
1080 457.25
1081 429.23
1082 447.19
 Ret
Ret
Compound Name K, Time MS
N-({1-[1 -{4-{[(2R)-2- hydroxypropyi]oxy}-2,3-
1083 dimethyiphenyl)ethyf]azet 1 16 425 23 idιn-3-yl}methyl)-2,5- dimethylbenzamide
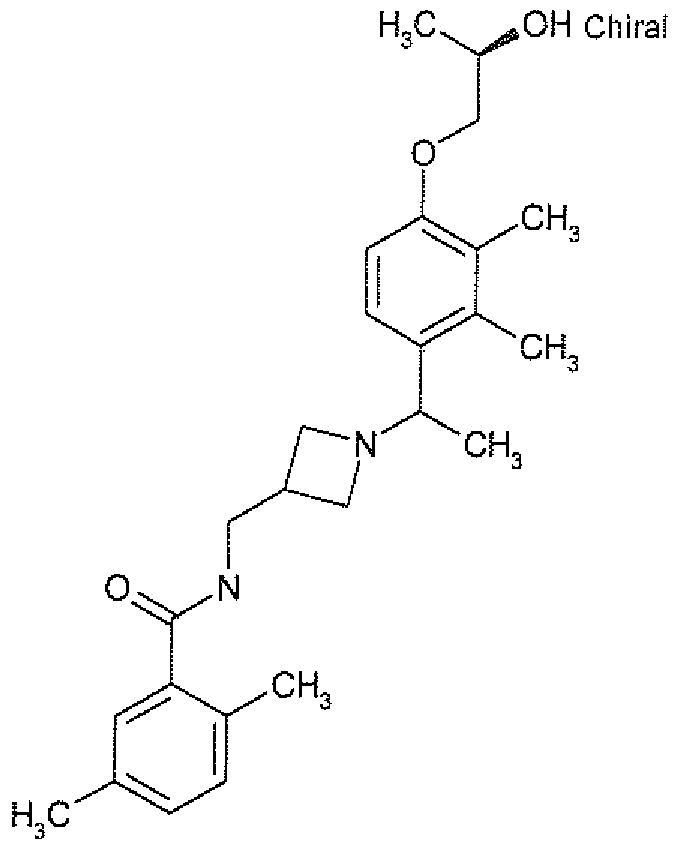
1084 * 1 07 402 29



1088 azet 116 47123
 Ret
Ret
Compound Name K, Time MS

1090 489 28
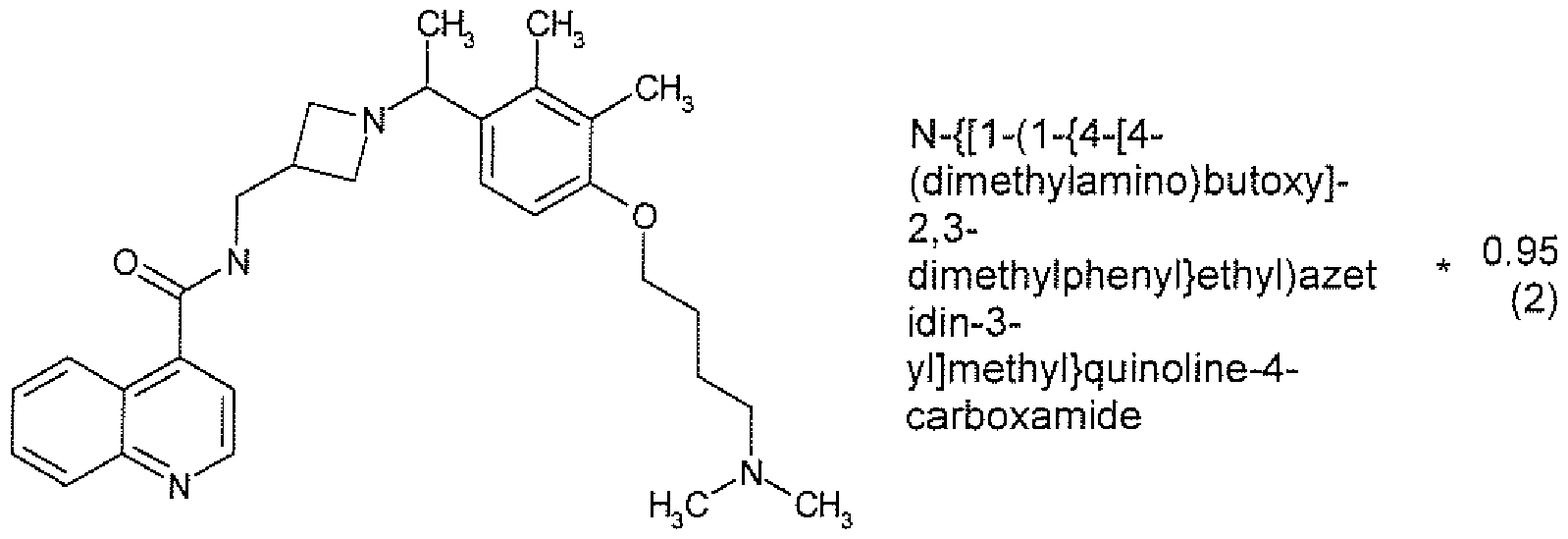

Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K, Time MS


 Ret
Ret
Compound Name K; Time MS
1105 et 1.15 429.21


Compound Name K,- Time MS
1 498.29

, 3-
1109 !}azet 1.15 441.23 (2)

no 417.27
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret

Compound Name K, Time MS
 Ret
Ret
Compound Name K, Time MS
 Ret
Ret

Ret
Compound Name K1 Time MS
1

Ret
Compound Name K, Time MS
2-fluoro-N-[<1-{1-[4-(4- hydroxybutoxy)-2,3-
1 130 dimethylpheny[]ethyl}azet 1.16 443.22 idin-3-yi)methyl]-5- methylbeπzamide

1 13 1 1.15 446.20 ide
1 132 1.08 460.23

Compound Name Ki Time MS
1333 1.12 535.24
1134 1.05 546.33

Ret
Compound Name K, Time MS
1136 zet * 117 43925
1137 44719
1138 45629
 Ret
Ret
Compound Name K, Time MS

1141 t 105 44623
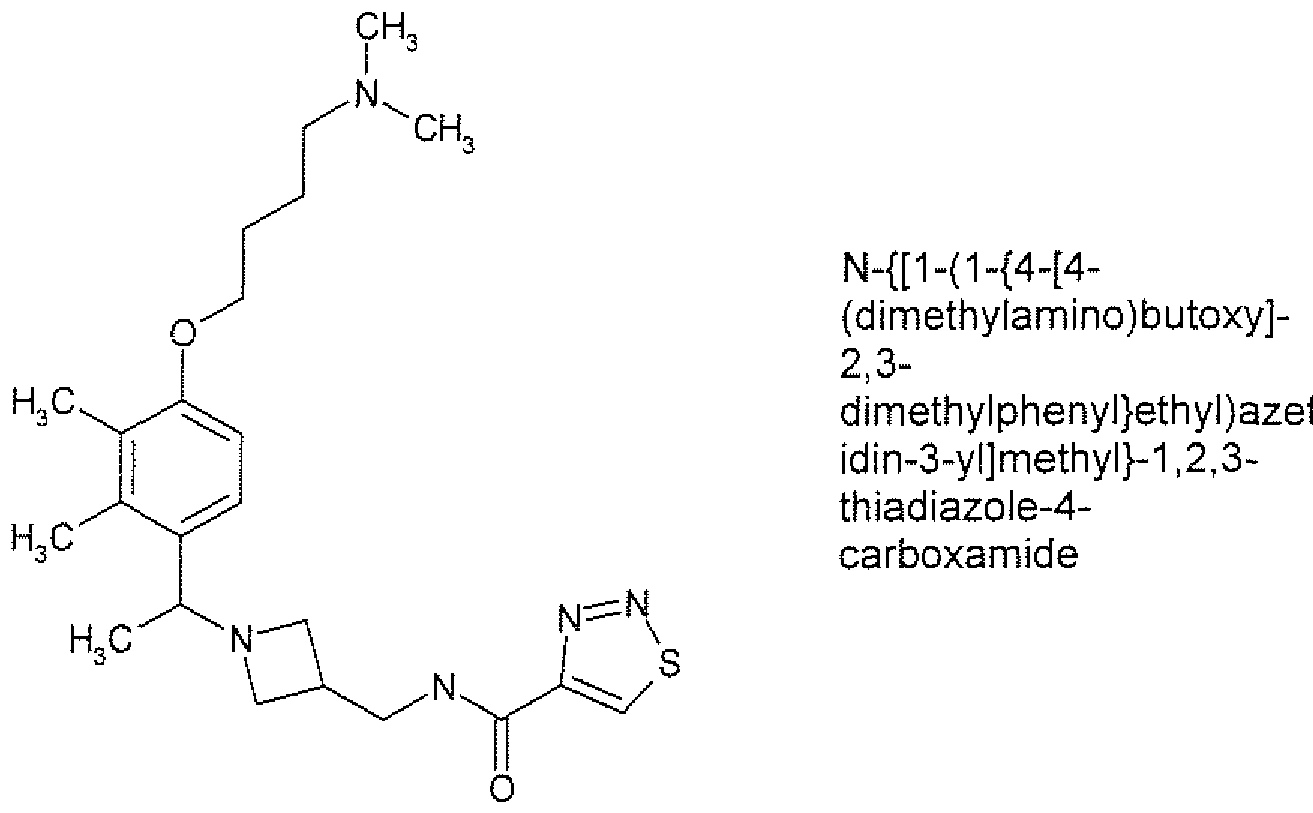 Ret
Ret
Compound Name K, Time MS
Ret

1147 43622
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K, Time MS
099 52826
107 45727
118 44322
 Ret
Ret
Compound Name K5 Time MS
 Ret
Ret
Compound Name K1 Time MS
106 42029
109 47712
108 50510
 Ret
Ret
Compound Name Kj Time MS

3-fluoro-N-[(1 -{1 -[4-(4- hydroxybutoxy)-2,3-
1 162 dimethylphenyl]ethyl}azet 1 16 443 22 ιdin-3-yl)methyl|-2- methyibenzamide


3-fluoro-N-[(1-{1-[4-{4- hydroxybutoxy}-2,3-
165 dimethylphenyi]ethyl}azet 1.15 459.21 idin-3-yl)methyl]-4- methoxybenzamtde

Compound Name K1 Time MS tert-butyl ({H1-(4-{[(2R)-
1166 dιmethy!pheπyl)ethyl]azet * 1 13 393 22 3 y ldl}mm-e3"thyl)carbamate


Compound Name K, Time MS
 Ret
Ret
Compound Name K1 Time MS
 Ret
Ret
Compound Name K1 Time MS
1176 114 42623
1177 094 522.24
1178 096 45328
 Ret
Ret
Compound Name Ks Time MS

1181 t 107 48731
1182 122 41336
 Ret
Ret
Compound Name K, Time MS

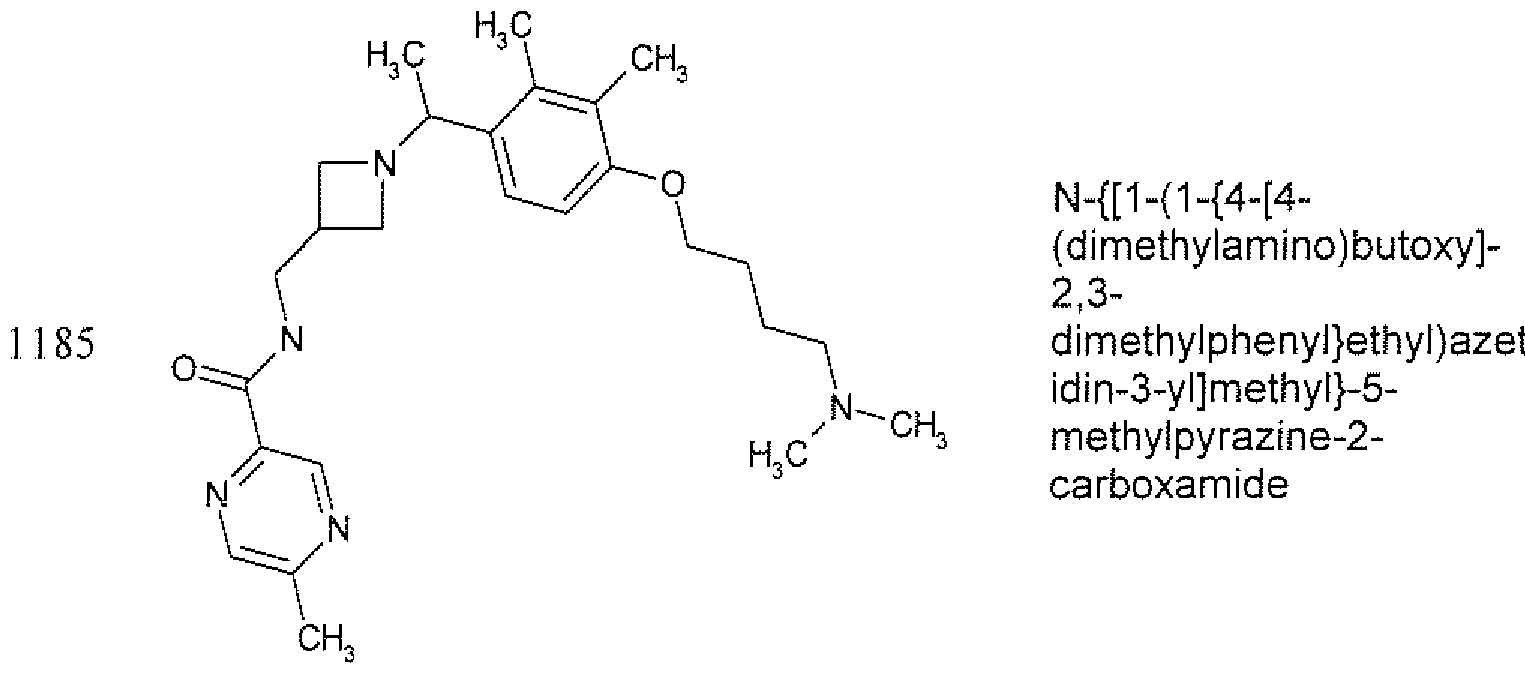
Compound Name K,- Time MS
 Ret
Ret
Compound Name Ki Time MS
1.13 507.24
* 1.14 393.24
* 104 432.29
1195 1.19 373.03

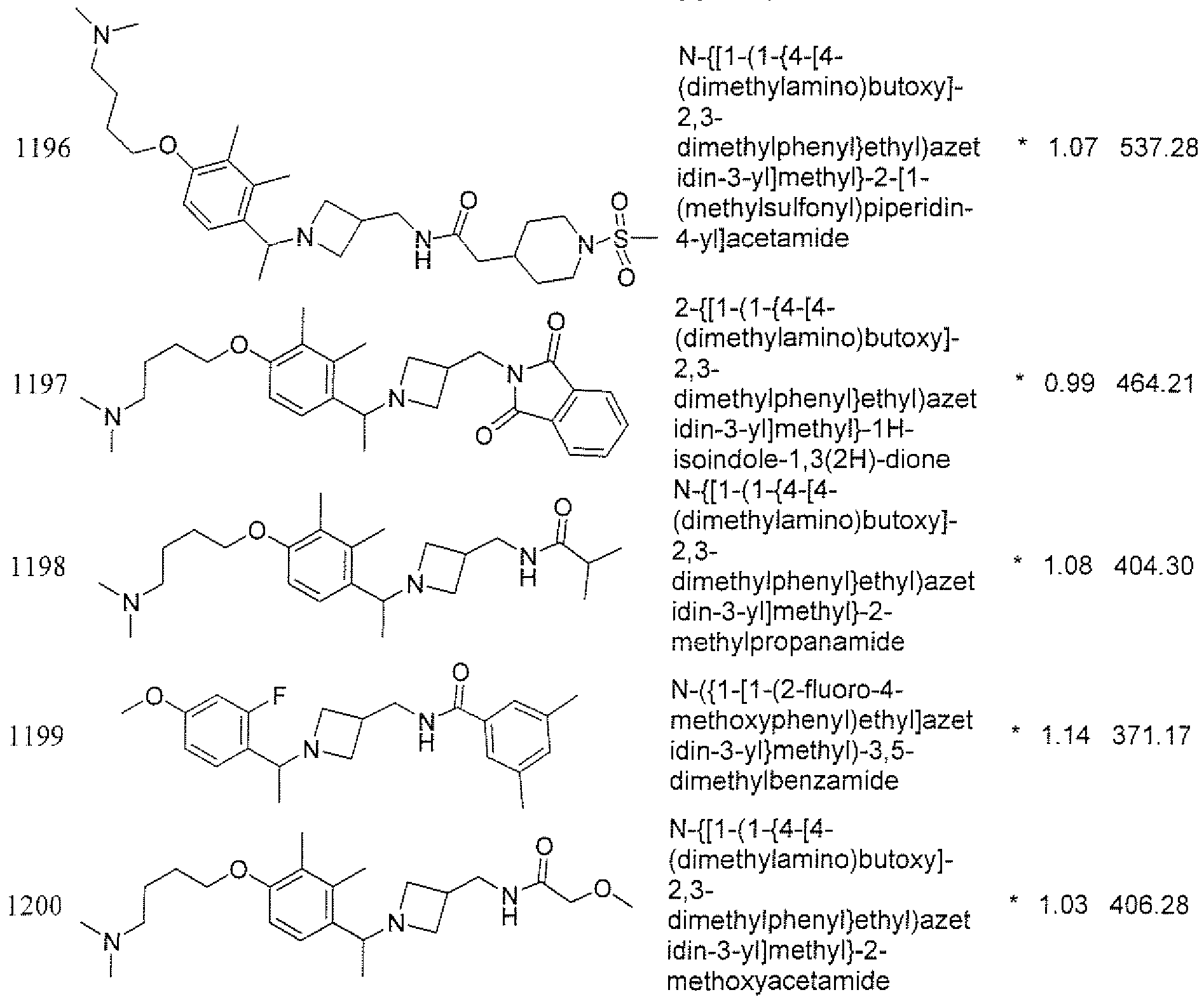
Compound Name K1 Time MS
 Ret
Ret

Ret
Compound Name Kj Time MS

Compound Name K, Time MS
2-chioro-N-{841-(2,3- dιmethyϊ-4-{[(2R}-5-
122<E oxopyrrolιdιn-2- * 1.08 511 33 yi]methoxy}phenyl)ethyi]-8- azabιcyclo[3 2 1]oct-3-
 yl}nicotinam!de
yl}nicotinam!de

N-{8-[1-(2,3-dimethyl-4- {[(2R)-5-oxopyrro(idin-2-
1232 yl]methoxy}phenyl)ethyl]-8- 1 04 491 37 azabιcyclo[3 2 1]oct-3-yl}-2- methylπtcotinamide



Compound Name K1 Time MS
N-{8-[1-(2,3-dιmethyl-4-
{[(2R}-5-oxopyrrolιdιn-2- yl]methoxy}phenyl)ethyl]-8- 1 12 491 37 azabicyclo[3 2 1]oct-3-yl}-6- methylpyπdιne-2- carboxamide
6-bromo-N-{8-[i-(2,3- dιmethyl-4-{[(2R}-5- oxopyrrohdm-2- 113 55725 y[]methoxy}phenyl)ethyl]-8- azabιcyclo[3 2 1]oct-3- ylJpyπdine-2-carboxamide
5,6-dιchloro-N-{8-{1-(2,3- dιmethy!-4-{[(2R)-5- oxopyrrolιdιn-2- 116 54526 yl3methoxy}phenyf)ethyl3-8- azabιcyclo[3 2 1]oct-3- yl}nιcotιnamιde
N-{8-[1-(2,3-dιmethyl-4-
{[(2R)-5-oxopyrrolidιn-2- yl]methoxy}phenyl)ethyt]-8- 110 49136 azabicyclo[3 2 1]oct-3-yl}-3- methylpyrιdine-2- carboxamide
N-{8-{1-(2,3-dιmethyl-4- {[(2R)-5-oxopyrrolidιn-2- y[jmethoxy}phenyl)ethyl]-8- 107 49136 azab!cyclc[3 2 1]oct-3-yl}-5- methylnicotinamide
NN--{{88--[[11--((22,,33--ddιimmeetthhyyll--4- {[(2R)-5-oxopyrrolid(n _2- yyllj]mmeetthhooxxyy}}pphheennyyll))eeUthy!]-8- 108 46733 azabιcyclo[3 2 1 Joct-3-yt}- 1 r3-oxazole-4-carboxamιde


3-chloro-N-{8-[1-(2,3- dιmethy!-4-{[(2R)-5- oxopyrrohdιπ-2- * 110 51130 S yJ]methoxy}phenyl)ethyl]-8- azabicyclo[3 2 1]oct-3-
 yljisoπicotiπamtde
Ret
yljisoπicotiπamtde
Ret
Name K1 Time MS
N-{8-[1-(2,3-dimethy!-4- {[(2R)-5-oxopyrrolidin-2-
125C yl]methoxy}phenyi)ethyl]-8- 1.05 491.36 azabicyclo[3.2.1 ]oct-3-yl}-3- methylisonicotinamide


N-{8-[1-{2,3-dtmethyl-4-
{[(2R)-5-oxopyrrolidin-2-
1254 yl]methoxy}phenyl)ethyi]-8- 1.14 521.35 azabicyclo[3.2.1 ]oct-3-yϊ}-2- ethoxyisonicotinamtde
N-{8-[1-(2,3-dimethyl-4-
{[(2R)-5-oxopyrroiidin-2- yl]methoxy}pheny[)ethyl]-8- 1.10 471.39
1255 azabicyclo[3.2.1 ]oct-3-yl}~
N'-methylpyrtdine-2,6-
 dicarboxamide
dicarboxamide


2-chloro-N-[8-(2,3-dιmethyl- 4-{[{2R)-5-oxopyrrolιdιπ-2- yl]methoxy}benzyl)-8- 1 09 497 31 azabιcyclo[3 2 1]oct-3- yφsonicotinamide
2-chloro-N-[8-(2 3-dιmethyl- 4-{[(2R)-5-oxopyrro!tdin-2- yl]methoxy}benzyl)-8- 1 10 511 31 azabιcyclo[3 2 1]oct-3-yl]-6- methyjisonicotinamide
2-chloro-N-[8-(2,3-dιmethyl- 4-{[(2 R)-5-oxopy rro lidi n-2- yl]methoxy}benzyl)-8- 1 14 527 32 azabιcyclo[3 2 1]oct-3-y!]-6- methoxyisonicotinamide
N-[8-(2,3-dιmethyl-4-{[(2R)-
5-oxopyrrolιdιn-2- yl]methoxy}benzy!)-8- 1 11 454 38 azab!cyclo[3 2 1]oct-3- yl]fsoxazole-5-carboxamιde
5-bromo-N-[8-(2,3-dιmethyl- 4-{[(2R)-5-oxopyrrolιdm-2- yl]methoxy}benzy!)-8- 1 12 543 25 azabscyclo[3 2 1]oct-3- yl]πιcotinamιde
 Ret
Ret
Name K, Time MS
N-[8-(2,3-dimethy[-4-{[(2R)- 5-oxo pyrrol id ιn-2-
1266 y!3methoxy}benzyl)-8- 1 04 477 37 azabιcyclo[3 2 1]oct-3-yl]-2- methylπicotinamide


N-[8-(2,3-dιmethyl-4-{[(2R)-
5-oxopyrrolιdm-2-
1272 yJ]methoxy}benzyl)-8- 1 07 491 38 azabιcyc(o[3 2 1]oct-3-yl]-5- ethylnicotinamide
N-[8-(2,3-dιmethyl-4-{[(2R)- 5-oxopyrrolιdιn-2-
1273 yl]methoxy}benzyl}-8- 1 15 494 37 azabιcyclo[3 2 1]oct-3-yl]-3- fluoro-5-methylbeπzamιde
 Ret
Ret
Compound Name K, Time MS

N-[8-(2,3-dιmethyl-4-{[{2R)- 5-oxopyrrolιdιn-2- yl]methoxy}benzyl)-8- 1 06 453 33 azabιcyclo[3 2 1]oct-3-yl]- 1 ,3-oxazole-4-carboxamιde
 Ret
Ret


111 52822

1
 Ret Ki Time MS -
Ret Ki Time MS -
1.10 538.28
-[4-
1.16 562.15 -[4-
1.14 562.16


1 1.10 558.21 -4-


Compound Name K1 Time MS

1 13 542 22 -4-
* 1 06 529 21


Compound Name K1 Time MS
2-(2,5-dιmethoxyphenyl)-N-
[(1 R)-8-{1-[4-({(2S)-3-
[ethyl(methyl)amιno]-2-
13 hydroxypropyi}oxy)-2,3- 1 07 568 28 dιmethyiphenyJ]ethyl}-8- azabιcyc!o[3 2 1]oct-3- yijacetamide
 -
-
1 07 572 24
 yljnicotinamtde
yljnicotinamtde
N-[(1 R)-8-{1-[4-({(2S)-3-
[ethyl(methyl)amιno]-2- hydroxypropyf}oxy)-2,3-
13 14 ' -ψ- dιmethyiphenyϊ]ethyf}-8- 1 05 486 31 azabιcycto[3 2 1]oct-3- yl]cyclopentanecarboxamid

N-[(1 R)-8-{1-[4-({(2S)-3-
[ethyl(methyl)amιno]-2- hydroxypropyl}oxy)-2,3- dιmethy[phenyl]ethyl}-8- 1 10 546 18 azabιcydo[3 2 1]oct-3-yl]-5-
(methylthfθ)thιopheπe-2- carboxamide
N-[(1 R)-8-{1-[4-({(2S)-3-
[ethyl(methyl)amtno]-2- hydroxypropyl}oxy}-2,3- d!methy!phenyl]ethyl}-8- 1 05 509 28 azabscyclo[3 2 1]oct-3-yl]-6-
 methylpyrιdιne-2- carboxamide
Ret
methylpyrιdιne-2- carboxamide
Ret
Compound Name K, Time MS
1.07 575.13


N-[(1 R)-8-{1-[4-({(2S)-3-
[ethyi(methyi)amino]-2- hydroxypropy[}oxy}-2,3- dimethylpheny[]ethyl}-8- 1.07 563.20 azabicycto[3.2.1 ]oct-3-yi]-6- {trifluoromethyi)nicotinamid
N-[(1 R)-8-{1-[4-({(2S)-3-
[ethyl(methy[)amino]-2- hydroxypropyl}oxy)-2,3- dimethylphenyl]ethyl}-8- 1.07 564.21 azabicyclo[3.2.1 ]oct-3-yl]-2-
(trifluorcmethyl)pyr!m!dine-
5-carboxamide
N-[{1 R)-8-{1-[4-{{(2S)-3-
[ethyl(methyl)amiπo]-2- hydroxypropyl}oxy)-2,3- dimethy[pheπyl]ethyi}-8- 1.09 563.21 azabicyclo[3.2.1 ]oct-3-yl]-5-
(trifluoromethyl}pyridine-2- carboxamide
 Ret
Ret
Compound Name K, Time MS

2-{2,5-dsmethylphenoxy)-N-
[(1 R)-8-{1-[4-({(2S)-3-
[ethyl{methy!)aιniπo]-2- hydroxypropyi}oxy)-2,3- 1 13 552 29 dιmethylphenyl]ethyl}-8-
 azabιcycfo[3 2 1]oct-3- yl]acetamιde
azabιcycfo[3 2 1]oct-3- yl]acetamιde

1 0 95 510 27

N-[{1R)-8-{1-[4-({{2S)-3- [ethyl(methyl)amtπo]-2- hydroxypropyl}oxy)-2,3- 0 97 485 24 dιmethylphenyl]ethy!}-8- azabιcyclo[3 2 1]oct-3-yl]-
1 ,3-oxazole-4-carboxamιde
N-[(1 R)-8-{1-[4-({(2S)-3-
[ethyl(methyl)amιno]-2- hydroxypropy!}oxy)-2,3- dιmethylpheπyl]ethyl}-8- 1 03 513 26 azabιcyclo[3 2 1]oct-3-yl]-
 2,4-dιmethy!-1 ,3-oxazole-5- carboxamide
2,4-dιmethy!-1 ,3-oxazole-5- carboxamide
1333 - 1 22 419 29 a
 Ret
Ret
Compound Name Kj Time MS

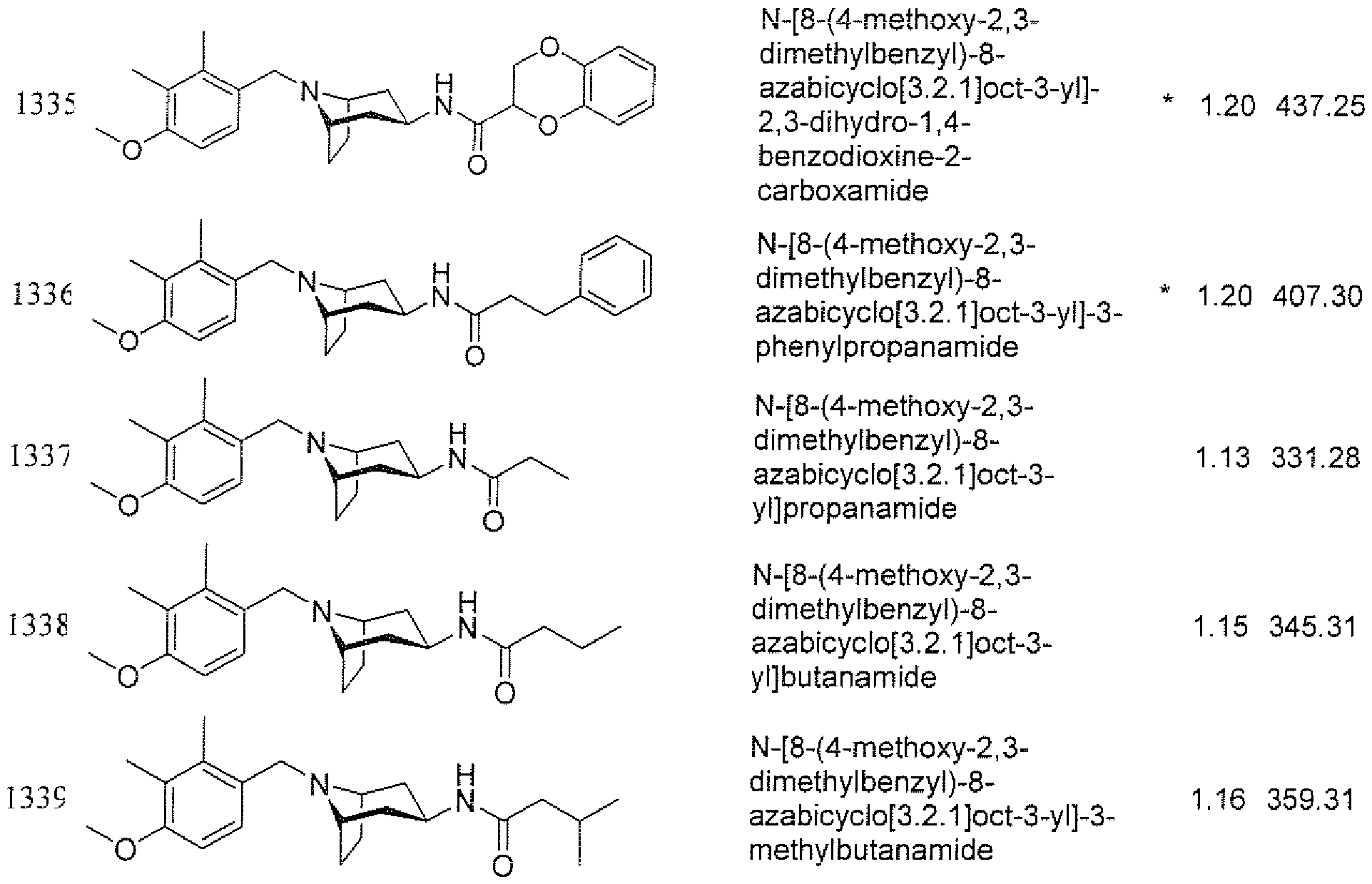
2-cyclopentyl-N-[8-{4- methoxy-2,3-
134C dimethylbenzyl)-8- 1.20 385.33 azabicyclo[3.2.1 Joct-3- yljacetamide
2-cyciohexyl-N-f8-{4- methoxy-2,3-
1341 dirnethylbenzyi)-8- 1.22 399.34 azabicyclo[3.2.1 ]oct-3-
 yljacetamide
yljacetamide

Compound Name K1 Time MS
1344 1.18 371.32 mid
1345 1.21 399.33 mid

N-[8-(4-methoxy-2,3- dimethyibenzyl)-8-
1346 azabicyclo(3.2.1 ]oct-3- 1.14 373.29 yl]tetrahydrofuran-2- carboxamide
N-[8-(4-methoxy-2,3- dimethylbenzyi)-8-
1347 azabicyc(o[3.2.1 ]oct-3- 1.13 373.29 y!]tetrahydrofuran-3-
 carboxamide
carboxamide

1351 - 1.21 399.33

1352 2- 1.23 399.34 a
1353 1 - 1.21 419.30 xa
 Ret
Ret
Compound Name K, Time MS

N-[8-(4-methoxy-2,3- dimethylbenzyl}-8-
136C azabιcyc!o[3 2 1]oct-3-y[]-2- 1 14 401 32 {tetrahydro-2H-pyran-4- yl)acetamide
N-[8-(4-methoxy-2,3- d:methylbenzyl}-8-
1361 azabιcyc!o[3 2 1]oct-3- 1 10 435 28 yl]tetrahydro-2H-thιopyran- 4-carboxamιde 1 ,1 -dioxide
N-[8-(4-methoxy-2,3- d!rπethylbeπzyl)-8-
1362 azabιcyc!o[3 2 1]oct-3-yl]-1- 1 12 464 29 (methy!su!fonyl)pipendine- 4-carboxamιde
N-[8-(4-methoxy-2,3- dιmethylbenzyl}-8-
1363 azabιcydo[3 2 1]oct-3-ylJ-1- 1 12 464 29 (methylsulfonyl)pιpeπdιne-
 3-carboxamιde
Ret
3-carboxamιde
Ret
Compound Name K1 Time MS
N-[8-{4-methoxy-2,3- clιmethy[benzyi)-8-
1364 azabicycio[3 2 1 ]oct-3-yl]-1 - 1 14 464 28 (methylsulfonyl)pfperιdine-
 2-carboxamide
2-carboxamide

Compound Name K1 Time MS
N-[8-(4-methoxy-2,3- drmethylbenzyl)-8- azabιcyclo[3 2 1 ]oct-3-yl]-2- 1 13 428 33 (2-oxopιpeπdιn-1- yl)propaπamιde
N-[8-(4-methoxy-2,3- dιmethylbenzyl)-8- azabιcyclo[3 2 1 ]oct-3-y!]-2- 1 12 414 33 (2-oxopιpeπdιn-1- yl)acetamιde
N-[8-(4-methoxy-2,3- dimethylbenzyl)-8- azabιcyclo[3 2 1 ]oct-3-yi]-2- 1 10 400 30 (2-oxopyrrolιdιn-1-
 yl)acetamιde
yl)acetamιde

Compound Name K, Time MS

5-(4-chloropheπyl)-N-[8-(4- methoxy-2,3-
1391 dimethylbeπzyi)-8- 1.29 493.21 azabicyclo[3.2.1 ]oct-3-yl]-2- methyi-3-furamide
N-[8-(4-methoxy-2,3- dimethylbeπzyi)-8-
1392 azabicyclo[3.2.1]oct-3-yl]- 1.12 398.25
3,5-dimethylisoxazo!e-4- carboxamide
N-[8-(4-methoxy-2,3- dimethyϊbenzyl)-8-
1393 azabicyclo[3.2.1]oct-3-yl]- 1.13 414.23 2,4-dimethy!-1 ,3-thiazole-5- carboxamide
N-[8-(4-methoxy-2,3- dimethylbenzyl)-8-
1394 azabicyclo[3.2.1]oct-3-yl]- 1.15 436.21 1 ,3-beπzothtazole-6-
 carboxamide
Ret
carboxamide
Ret
Compound Name K1 Time MS

N-[8-(4-methoxy-2,3- dimethy!benzyl)-8-
1396 azabιcyclo[3 2 1]oct-3-yl]-5- 1 13 395 26 methyipyrazιne-2- carboxamide
N-[8-(4-methoxy-2,3- dimethylbenzyl)-8-
1397 azabιcyclo[3 2 1]oct-3-yl]-5- 1 18 459 28 methyl-1-phenyl-1 H- pyrazo[e-4-carboxamιde
N-[8-(4-methoxy-2,3- dimethyJbenzyl)-8-
1398 azabιcyclo[3 2 1 Joct-3-yl]-2- 1 14 463 22 pyπdιn-3-yM ,3-thιazole-4- carboxamide
N-[8-(4-methoxy-2,3- dimethylbenzyl)-8- azabιcyclo[3 2 1]oct-3-yl]-5-
1395 [1-methy!-3- 1 23 533 18
(tπf!uoromethyl)-1 H-pyrazol-
 5-yi]thιophene-2- carboxamide
5-yi]thιophene-2- carboxamide

26
26

N-[8-(4-methoxy-2,3- dιmethylbenzyl)-8-
1408 azabιcycio[3 2 1 Joct-3-y I]- 1 18 420 23 2,1-benzisoxazole-3-
 carboxamide
carboxamide

Compound Name K, Time MS

6-bromo-N~(8-{1-[4-(2- methoxyethoxy)-2,3- dsmethylphenyJ]ethyl}-8- * 1 23 516 18 azabιcyclo[3 2 1]oct-3- yl)pyrιdine-2-carboxamιde
3,6-dιchioro-N-(8-{1-[4-(2- methoxyethoxy)-2,3- dιmethylpheπyl3ethy[}-8- * 1 28 516 31 azabιcycb[3 2 1]oct-3-
 yl)pyπdιne-2-carboxamιde
yl)pyπdιne-2-carboxamιde

6-ethyl-N-(8-{1-[4-(2- methoxyethoxy}-2,3- dιmethylphenyl]ethyi}-8- 1 17 466 37 azabιcyclo[3 2 1]oct-3-
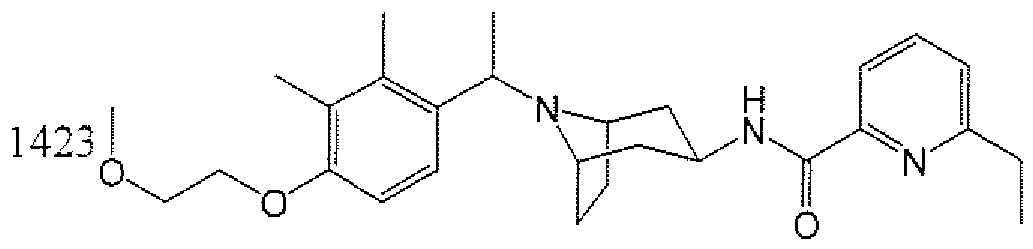 yi)pyπdιne-2-carboxamιde
yi)pyπdιne-2-carboxamιde

Compound Name K, Time MS
 Ret
Ret
Compound Name K1 Time MS

* 1.17 523.43


)- * 1 ,24 506.27
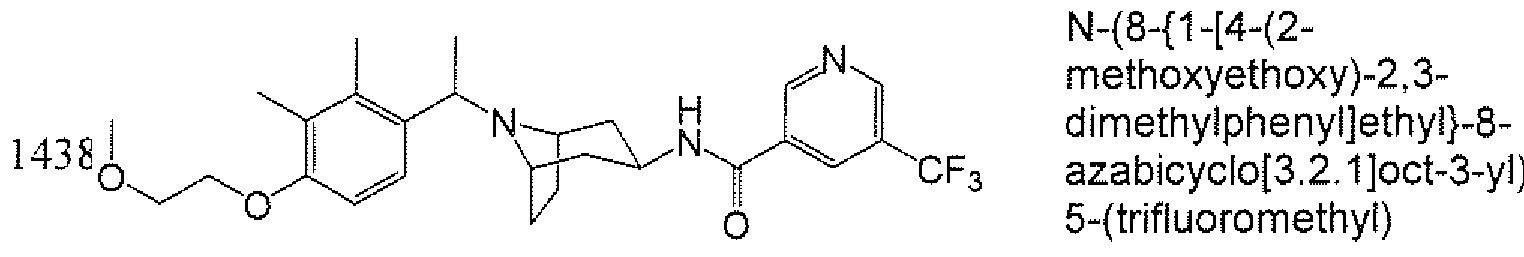 nicotinamide
nicotinamide
5-ethyny!-N-(8-{1-[4-(2- methoxyethoxy)-2,3- dimethy!phenyl]ethyl}-8- * 1 ,24 462,30 azabicycio[3.2.1]oct-3- yl)nicotinamide
6-ethyπyl-N-(8-{1-[4-(2- methoxyethoxy)-2, 3- dimethylpheny[]ethyl}-8- * 1.26 462.30 azabicyclo[3.2.1]oct-3-
 yl)pyridine-2-carboxamide
yl)pyridine-2-carboxamide
EXAMPLE 5. PURIFIED RA r STRIATUM CELL MEMBRANES.
The MCHlR receptor source is a rat striatum homogenate. The rats are naϊve Sprague Dawley or Wistar rats which are not food deprived overnight, and weigh roughly 250±25 grams. The striatum is rapidly and carefully dissected away from the cortex, mid-brain and hippocampus. The striatum is weighed, and homogenized in Prep buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 2 mM EGTA: 23 mL per gram of striatum, typically 150 mg of tissue plus 3.5 mL of prep buffer), homogenizing for 30 seconds using a BRTNKMAN POLYTRON at setting 5. The crude striatal homogenate is washed 2 times with Prep buffer and sampled for protein analysis between washes. Once the protein concentration has been determined, the final protein pellet is suspended in binding buffer at a protein density of 275 μg / 200 μL binding buffer. The protein concentration of the resulting membrane preparation (hereinafter Mrat striatal membranes") is conveniently measured using a Bradford protein assay (Bio-Rad Laboratories, Hercules, CA).
EXAMPLE 6. RADIOLIGAND BINDING ASSAYS
This Example illustrates a standard assay of Melanin Concentrating Hormone receptor binding that may be used to determine the binding affinity of compounds for the MCH receptor. l23I~labeled S36057 (New England Nuclear Corp., Boston, MA), a stable analogue of MCH, is used as the radioligand.
Purified rat striatal membranes, prepared by the method given above, are resuspended by Dounce homogenization (tight pestle) in binding buffer (50 mM Tris pH. 7.4, 1.0 mM Mg Cl2, 5 mM KCl, 1 mM CaCl2, 120 mM NaCI, 1 mM bacitracin, 0.02 mg/mL Aprotinin & 0.1% BSA).
The optimal rat striatal homogenate input has been determined, via a protein linearity experiment, to be 275 μg / data point / 250 μL. At 3OpM [i25I]-S36057, this amount of protein binds 10-15% of the input radioligand. At a [125I]-S36057 input of 30 pM (roughly 1/2 to 1/3 Kd) the specific binding signal is routinely 50%. Nonspecific binding is defined with iμM MCH. Displacement binding studies, designed to deteπnine the IC50 and K1 of exogenousiy added compounds, are performed using 30 pM [l25I]-S36057. These displacement studies are routinely run to verify activity in the rat striatum homogenate MCHlR preparation. Upon mixing of all assay components (100 μL tissue, lOOμl assay buffer, 25 μL radioiabel, and 2.5 μL test compound if required, 25 μL binding buffer), the reaction is mixed and incubated at RT for 2 h in a 96-well deepwell dish. The binding reaction is terminated by rapid filtration over a 1% PEI treated filter on a 96-well Tomtec harvester, followed by washing with 50 mM Tris, pH 7.4, 120 mM NaCl. For saturation binding analysis, rat striatal membranes (275 μg) are added to polypropylene tubes containing 25 pM - 0.5 nM [!25I]S36057. Nonspecific binding is determined in the presence of 10 μM MCH (Tocris Cookson Inc., Ellisville, MO, USA) and accounts for less than 10 % of total binding. For evaluation of guanine nucleotide effects on receptor affinity, GTPγS is added to duplicate tubes at the final concentration of 50 μM. For competition analysis, membranes (275 μg) are added to polypropylene tubes containing
0.03 nM [U3I]S36057. Non-radiolabeled displacers are added to separate assays at concentrations ranging from 10"10 M to 10"5 M to yield a final volume of 0.250 mL. Nonspecific binding is determined in the presence of 10 μM MCH and accounts for less than 30% of total binding. Following a 2 hour incubation at rt, the reaction is terminated by rapid vacuum filtration. Samples are filtered over presoaked (0.3% non-fat dry milk for 2 h prior to use) GF/C WHATMAN filters and rinsed 2 times with 5 mL cold 50 mM Tris pH 7.4. Remaining bound radioactivity is quantified by gamma counting. K, and Hill coefficient ("nH") are determined by fitting the Hill equation to the measured values with the aid of SIGMAPLOT software.
EXAMPLE 7. PURIFIED RECOMBINANT CHO CELL MEMBRANES EXPRESSING MONKEY MCHlR Cynomolgus macaque hypothalamus MCHl R cDNA is prepared and cloned into
PCDNA3.1 (INVITROGEN Corp., Carlsbad, CA) as described in PCT International Application
publication number WO 03/059289, which published on July 24, 2003. The resulting MCHl expression vector is stably transfected into Chinese hamster ovary (CHO) cells (American Type Culture Collection, Manassas, VA) via calcium precipitation. The disclosure of WO 03/059289 at page 51-52 directed to the preparation and storage of membrane pellets prepared from CHO ceils stably transfected with the MCHl vector is hereby incoiporated by reference.
CHO mMCHlR cell pellets are resuspended in homogenization buffer (10 inM HEPES, 250 mM sucrose, 0.5 μg/mL leupeptin, 2 μg/mL Aprotinin, 200 μM PMSF, and 2.5 mM EDTA, pH 7.4) and homogenized using a BRINKMAN POLYTRON homogenizer (setting 5 for 30 seconds). The homogenate is centrifuged (536 x g/ 10 min/ 40C) to pellet the nuclei. The supernatant containing isolated membranes is decanted to a clean centrifuge tube, centrifuged (48,000 X g/ 30 min. 4°C) and the resulting pellet resuspended in 30 mL homogenization buffer. This centrifugation and resuspension step is repeated twice. The final pellet is resuspended in ice cold Dulbecco's PBS containing 5 mM EDTA and stored in frozen aiiquots at -8O0C until needed. The protein concentration of the resulting membrane preparation (hereinafter "P2 membranes") is conveniently measured using a Bradford protein assay (Bio-Rad Laboratories, Hercules, CA).
EXAMPLE 8. AGONIST-INDUCED GTP BINDING
Agonist-stimulated GTP gamma3=S binding ("GTP binding") activity can be used to identify agonist and antagonist compounds and to differentiate neutral antagonist compounds from those that possess inverse agonist activity. This activity can also be used to detect partial agonism mediated by antagonist compounds. A compound being analyzed in this assay is referred to herein as a "test compound."
Agonist-stimulated GTP binding on purified P2 membranes (prepared as described above) is assessed using MCH as agonist in order to ascertain the level of signal, and EC50 value of MCH as measured by GTP binding. P2 membranes from the CHO cells are resuspended by Dounce homogenization (tight pestle) in GTP binding assay buffer (50 mM Tris pH 7.4, 120 mM NaCl, 5 mM MgC12, 2 mM EGTA, 0.1% BSA, 0.1 mM bacitracin, 100 KIU/mL aprotinin, 5 μM GDP, 10 μg/mL saponin) and added to reaction tubes at a concentration of 50 μg protein/reaction tube. After adding increasing doses of the agonist MCH at concentrations ranging from 10"12 M to 10"6 M, reactions are initiated by the addition of 100 pM GTP gamma~5S. In competition experiments, non-radio labeled test compounds (e.g., compounds provided herein) are added to separate assays at concentrations ranging from 10"10 M to 10"5 M along with 10 nM MCH to yield a final volume of 0.25 mL.
Neutral antagonists are those test compounds that reduce the MCH stimulated GTP binding activity towards, but not below, baseline (the level of GTP bound by membranes in this assay in the absence of added MCH or other agonist and in the further absence of any test compound).
An antagonist test compound that elevates GTP binding activity above baseline in the absence of added MCH in this GTP binding assay is characterized as having partial agonist activity.
Preferred antagonist compounds described herein do not elevate GTP binding activity under such conditions more than 10% above baseline, preferably not more than 5% above baseline, and most preferably not more than 2% above baseline.
Following a όθ-min incubation at rt, the reactions are terminated by vacuum filtration over GF/C filters (pre-soaked in wash buffer, 0.1% BSA) followed by washing with ice-cold wash buffer (50 niM Tris pH 7.4, 120 mM NaC!). The amount of G-alpha-bound (and thereby membrane- bound) GTP gamma35S is determined by measuring the bound radioactivity, preferably by liquid scintillation spectrometry of the washed filters. Non-specific binding is determined using 10 mM GTP gamma35S and typically represents less than 10% of total binding. Data is expressed as percent above basal (baseline). The results of these GTP binding experiments are analyzed using SlGMAPLOT software and IC50 determined. The IC50 is then used to generate K1 as described by Cheng and Prusoff (1973) Biochem Pharmacol 22(23):3099-\ 08. Preferred compounds are MCHI receptor antagonists that do not possess significant (e.g., greater than 5%) agonist activity in any of the MCH mediated functional assays discussed herein. Specifically, this undesired agonist activity can be evaluated, for example, in the GTP binding assay described above, by measuring small molecule mediated GTP binding in the absence of the agonist, MCH. The preferred extent of MCHlR agonist activity exhibited by compounds of the invention is less than 10%, more preferably less than 5% and most preferably less than 2% of the response elicited by the agonist, MCH.
EXAMPLE 9. MELANIN CONCENTRATING HORMONE RECEPTOR BINDING ASSAY
This Example illustrates a standard assay of melanin concentrating hormone receptor binding that may be used to determine the binding affinity of compounds for the MCH receptor.
Cynomolgus macaque hypothalamus MCHlR cDNA is prepared and cloned into PCDNA3.1 (ΪNVΪTROGEN Corp., Carlsbad, CA), and HEK293 cells (American Type Culture Collection, Manassas, VA) are stably transfected with the MCHl expression vector as described in PCT International Application publication number WO 03/059289, which published on July 24, 2003. The disclosure of WO 03/059289 at page 52 directed to the preparation and storage of the transfected HEK293 cells is hereby incorporated by reference.
At the time of assay, pellets are thawed by addition of wash buffer (25 mM HEPES with 1.0 mM CaCl2, 5.0 mM MgCl2, 120 mM NaCl, pH 7.4) and homogenized for 30 seconds using a BRINKMAN POLYTRON, setting 5. Cells are centrifuged for 10 min at 48,000 x g. The supernatant is discarded and the pellet is resuspended in fresh wash buffer, and homogenized again. An aliquot of this membrane homogenate is used to determine protein concentration via the Bradford method (BIO-RAD Protein Assay Kit, #500-0001, BlO-RAD, Hercules, CA). By this
measure, a 1 -liter culture of cells typically yields 50-75 mg of total membrane protein. The homogenate is centrifuged as before and resuspended to a protein concentration of 333 μg/mL in binding buffer (Wash buffer + 0.1% BSA and 1.0 μM final phosphoramidon) for an assay volume of 50 μg membrane protein/150 μl binding buffer. Phosphoramidon is from SIGMA BIOCHEMICALS, St. Louis, MO (cat# R-7385).
Competition binding assays are perfoπned at rt in Falcon 96 well round bottom polypropylene plates. Each assay well contains 150 μL of MCH receptor-containing membranes prepared as described above, 50 μL i25l-Tyr MCH, 50 μL binding buffer, and 2 μL test compound in
DMSO. 125!-Tyr MCH (specific activity = 2200 Ci/mmol) is purchased from NEN, Boston, MA (Cat # NEX 373) and is diluted in binding buffer to provide a final assay concentration of 30 pM.
Non-specific binding is defined as the binding measured in the presence of 1 μM unlabeled MCH. MCH is purchased from BACHEM U.S.A., King of Prussia, PA (cat # H-1482). Assay wells used to deteπnine MCH binding contain 150 μL of MCH receptor containing membranes, 50 μL !25I-Tyr MCH, 25 μL binding buffer and 25 μL binding buffer. Assay plates are incubated for 1 h at room temperature. Membranes are harvested onto
WALLAC™ glass fiber filters (PERKIN-ELMER, Gaithersburg, MD) which are pre-soaked with 1.0% PEl (polyethyleneimine) for 2 h prior to use. Filters are allowed to dry overnight, and then counted in a WALLAC 1205 BETA PLATE counter after addition of WALLAC BETA SCINT™ scintillation fluid. For saturation binding, the concentration of i25I-Tyr MCH is varied from 7 to 1,000 pM.
Typically, 11 concentration points are collected per saturation binding curve. Equilibrium binding parameters are determined by fitting the allosteric Hill equation to the measured values with the aid of the computer program FitP™ (BlOSOFT, Ferguson, MO). For preferred compounds, Kj values are below 1 micro molar, preferably below 500 nanomolar, more preferably below 100 nanomolar.
EXAMPLE 10. CALCIUM MOBILIZATION ASSAY
This Example illustrates a representative functional assay for monitoring the response of cells expressing melanin concentrating hormone receptors to melanin concentrating hormone. This assay can also be used to determine if test compounds act as agonists or antagonists of melanin concentrating hormone receptors.
Chinese Hamster Ovary (CHO) cells (American Type Culture Collection; Manassas, VA) are stably transfected with the MCH expression vector via calcium phosphate precipitation, and are grown to a density of 15,000 cells/well in FALCON™ black-walled, clear-bottomed 96-well plates (#3904, BECTON-DICKINSON, Franklin Lakes, NJ) in Ham's F12 culture medium (MEDIATECH, Herndon, VA) supplemented with 10% fetal bovine serum, 25 inM HEPES and 500 μg/mL (active) G418. Prior to running the assay, the culture medium is emptied from the 96 well
plates. Fluo-3 calcium sensitive dye (Molecular Probes, Eugene, OR) is added to each well (dye solution: 1 mg FLUO-3 AM, 440 μL DMSO and 440 μL 20% pluronic acid in DMSO, diluted 1 :4, 50 μL diluted solution per well). Plates are covered with aluminum foil and incubated at 370C for 1- 2 h. After the incubation, the dye is emptied from the plates, cells are washed once in 100 μL KRH buffer (0.05 niM KCl, 0.1 15 M NaCl, 9.6 mM NaH2PO4, 0.01 mM MgSO4, 25 mM HEPES, pH 7.4) to remove excess dye; after washing, 80 μL KRH buffer is added to each well.
Fluorescence response is monitored upon the addition of either human MCH receptor or test compound by a FLIPR™ plate reader (Molecular Devices, Sunnyvale, CA) by excitation at 480 nm and emission at 530 nm. In order to measure the ability of a test compound to antagonize the response of cells expressing MCH receptors to MCH, the EC50 of MCH is first determined. An additional 20 μL of KRH buffer and 1 μL DMSO is added to each well of cells, prepared as described above. 100 μL human MCH in KRH buffer is automatically transferred by the FLIPR instrument to each well. An 8-point concentration response curve, with final MCH concentrations of 1 nM to 3 μM, is used to determine MCH EC50.
Test compounds are dissolved in DMSO, diluted in 20 μL KRH buffer, and added to cells prepared as described above. The 96 well plates containing prepared ceils and test compounds are incubated in the dark, at room temperature for 0.5-6 h. It is important that the incubation not continue beyond 6 h. Just prior to determining the fluorescence response, 100 μL human MCH diluted in KRH buffer to 2 x EC50 is automatically added by the FLIPR instrument to each well of the 96 well plate for a final sample volume of 200 μL and a final MCH concentration Of EC50. The final concentration of test compounds in the assay wells is between 1 nM and 5 μM. Typically, cells exposed to one EC5O of MCH exhibit a fluorescence response of about 10,000 Relative
Fluorescence Units. Cells incubated with antagonists of the MCH receptor exhibit a response that is significantly less than that of the control ceils to the p<0.05 level, as measured using a parametric test of statistical significance. Typically, antagonists of the MCH receptor decrease the fluorescence response by about 20%, preferably by about 50%, and most preferably by at least 80% as compared to matched controls. IC50 values for MCHR antagonists are deteπnined using SIGMAPLOT software (SPSS Inc., Chicago, IL) and standard techniques. The IC50 is then used to generate K, as described by Cheng and Prusoff (1973) Biochem Pharmacol. 22(23)-3099A0%.
Alternatively, the data is analyzed as follows. First, the average maximum relative fluorescent unit (RFU) response from negative control wells (no agonist) is subtracted from the maximum response detected for each of the other experimental wells. Second, average maximum
RFU response is calculated for the positive control wells (agonist wells). Then, percent inhibition for each compound tested is calculated using the equation:
n * r L -U V mn i nn f Peak Signal in Test Wells 1
Percent Inhibition - 100 - 100 x ~ — ; — : — 65T^ — 7 — -—-—
L Peak signal in Agonist Wells J
The % inhibition data is plotted as a function of test compound concentration and test compound IC50 is determined using a linear regression in which x is ln(concentration of test compound) and y is ln(percent inhibition/(100 - percent inhibition). Data with a percent inhibition that is greater than 90% or less than 15% are rejected and are not used in the regression. The IC50 is eC-tøα∞pMopή and js useci t0 generate Kb as described above.
The ability of a compound to act as an agonist of the MCH receptor is determined by measuring the fluorescence response of cells expressing MCH receptors, using the methods described above, in the absence of MCH. Compounds that cause ceils to exhibit fluorescence above background are MCH receptor agonists (background autofluorescence of the test compound may be assessed using standard methods). Compounds that induce no detectable increase in the basal activity of the MCH receptor have no detectable agonist activity and are preferred.
EXAMPLE l L MDCK CYTOTOXICITY ASSAY
This Example illustrates the evaluation of compound toxicity using a Madin Darby canine kidney (MDCK) cell cytotoxicity assay. 1 μL of test compound is added to each well of a clear bottom 96-well plate (PACKARD,
Meriden, CT) to give final concentration of compound in the assay of 10 μM, 100 μM or 200 μM. Solvent without test compound is added to control wells.
MDCK cells, ATCC no. CCL-34 (American Type Culture Collection, Manassas, VA)5 are maintained in sterile conditions following the instructions in the ATCC production information sheet. Confluent MDCK cells are trypsinized, harvested, and diluted to a concentration of 0.1 x 1 06 cells/mL with warm (37°C) medium (VlTACELL Minimum Essential Medium Eagle, ATCC catalog # 30-2003). 100 μL of diluted cells is added to each well, except for five standard curve control wells that contain 100 μL of warm medium without cells. The plate is then incubated at 370C under 95% O2, 5% CO2 for 2 h with constant shaking. After incubation, 50 μL of mammalian cell lysis solution (from the PACKARD (Meriden, CT) ATP-LITE-M Luminescent ATP detection kit) is added per well, the wells are covered with PACKARD TOPSEAL stickers, and plates are shaken at approximately 700 rpm on a suitable shaker for 2 min.
Compounds causing toxicity will decrease ATP production, relative to untreated cells. The ATP-LITE-M Luminescent ATP detection kit is generally used according to the manufacturer's instructions to measure ATP production in treated and untreated MDCK cells. PACKARD ATP LITE-M reagents are allowed to equilibrate to room temperature. Once equilibrated, the lyoph.il ized substrate solution is reconstituted in 5.5 mL of substrate buffer solution (from kit). Lyophilized ATP standard solution is reconstituted in deionized water to give a 10 mM stock. For the five control wells, 10 μL of serially diluted PACKARD standard is added to each of the standard curve
control wells to yield a final concentration in each subsequent well of 200 nM, 100 nM, 50 nM. 25 nM and 12.5 nM. PACKARD substrate solution (50 μL) is added to all wells, which are then covered, and the plates are shaken at approximately 700 rpm on a suitable shaker for 2 min. A white
PACKARD sticker is attached to the bottom of each plate and samples are dark adapted by wrapping plates in foil and placing in the dark for 10 min. Luminescence is then measured at 22°C using a luminescence counter (e.g., PACKARD TOPCOUNT Microplate Scintillation and
Luminescence Counter or TECAN SPECTRAFLUOR PLUS), and ATP levels calculated from the standard curve. ATP levels in celis treated with test comρound(s) are compared to the levels determined for untreated cells. Cells treated with 10 μM of a preferred test compound exhibit ATP levels that are at least 80%, preferably at least 90%, of the untreated cells. When a 100 μM concentration of the test compound is used, cells treated with preferred test compounds exhibit ATP levels that are at least 50%, preferably at least 80%, of the ATP levels detected in untreated ceils.
EXAMPLE 12. MICROSOMAL IN VITRO HALF-LIFE
This Example illustrates the evaluation of compound half-life values (ti/2 values) using a representative iiver microsomal half-life assay.
Pooled human liver microsomes are obtained from XenoTech LLC (Kansas City, KS). Such iiver microsomes may also be obtained from In Vitro Technologies (Baltimore, MD) or Tissue Transformation Technologies (Edison, NJ). Six test reactions are prepared, each containing 25 μL microsomes, 5 μL of a 100 μM solution of test compound, and 399 μL 0.1 M phosphate buffer (19 mL 0.1 M NaH2PO4, 81 mL 0.1 M Na2HPO4, adjusted to pH 7.4 with H3PO4). A seventh reaction is prepared as a positive control containing 25 μL microsomes, 399 μL 0.1 M phosphate buffer, and 5 μL of a 100 μM solution of a compound with known metabolic properties (e.g., DIAZEPAM or CLOZAPINE). Reactions are preincubated at 39°C for 10 min.
Cofactor mixture is prepared by diluting 16.2 mg NADP and 45.4 mg glucose-6-phosphate in 4 mL 100 mM MgCl3. G!ucose-6-phosphate dehydrogenase solution is prepared by diluting
2Ϊ4.3 μL glucose-6-phosphate dehydrogenase suspension (Roche Molecular Biocherøicals;
Indianapolis, TN) into 1285.7 μL distilled water. 71 μL of starting reaction mixture (3 mL cofactor mixture; 1.2 mL glucose-6-phosphate dehydrogenase solution) is added to 5 of the 6 test reactions and to the positive control. 71 μL 100 mM MgCl2 is added to the sixth test reaction, which is used as a negative control. At each time point (0, 1, 3, 5 and 10 min), 75 μL of each reaction mix is pipetted into a well of a 96-well deep-well plate containing 75 μL ice-cold acetonitriie. Samples are vortexed and centrifuged 10 min at 3500 rpm (Sorval T 6000D centrifuge, HlOOOB rotor). 75 μL of supernatant from each reaction is transferred to a well of a 96-well plate containing 150 μL of a 0.5 μM solution of a compound with a known LC/MS profile (internal standard) per well. LC/MS analysis of each sample is carried out and the amount of unmetabolized test compound is measured as AUC, compound concentration vs. time is plotted, and the tm value of the test compound is
extrapolated. Preferred compounds provided herein exhibit in vitro t\a values of greater than 10 min and less than 4 h, preferably between 30 min and 1 h, in human liver microsomes.
From the foregoing it will be appreciated that, although specific embodiments have been described herein for purposes of illustration, various modifications may be made without deviating from the spirit and scope of the invention.
Claims
1. A compound of the Formula:

X is C(=O), SO2, C(O)N(Rn), C(O)-C(Rn)=C(R12), or C(=0)-[C(RnR13)]v(Y)u; wherein Y is O or S; u is O or 1 ; v is 1 or 2; and Rn and R12 are independently H, Ci-C6alkyl, CrC6a!koxy, C2- C6a]ky] ether, or taken together with a RB moiety to form a Cs-Cjcycloaikyl or a 5- to 7- membered heterocycloalkyl;
Z is O, NR14, C(=0)N(Ri4) Or N(R14)C(O), wherein R14 is H or CrC6alkyl;
RA is 6- to 10-membered aryl, 5- to 10-membered heteroaryl, Q-Cgalkyl, Cj-Cehaloalkyl, (C3- C8cycloalky!)C0-C4alkyl or (5- to 7-membered heterocycloalkyl)Co-C4alkyi, each of which is substituted with from O to 4 substituents independently chosen from RB; Each R3 is independently:
(i) halogen, hydroxy, nitro, cyano, amino, oxo, -COOH, aminocarbonyl or aminosulfonyl;
(ii) CpQalkyl, C2-C6alkenyl, C2-C6alkynyl, CrC6alkoxy, CrC6alkanoyL CrC6alkyithio, C1-
Cδalkanoyloxy, Cj-Qalkoxycarbonyl, Cj-Cgalkylsulfonyl, Cj-Cehaloalkyl, mono- or di-(Cr
C6a!kyl)aminoCo-C4alkyl, mono- or di-(C1-C6alkyl)aminocarbonylCo-C4alkyI, mono- or di-
(Ci-C6alkyl)aminosu]fonyICo-C4alkyl, (C3-C7cycloalkyI)C0-C2alkyl, (4- to 10-membered heterocycIe)C0-C2alkyl, phenylC0-C2alkyl or (3- to 10-membered cycle)C0-C2alkoxy; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CrC4alkyl, C]-C4alkoxy, CrQhaloalkyl, CrC4haloalkoxy, mono- or di-(CrC6alkyl)amino, or 5- or 6-membered heterocycloalkyi; or
(iii) taken together with a Rn or R52 moiety to form a C5-C7 eye loalkyl or a 5- to 7-membered heterocycloalkyi; or two adjacent RB groups are taken together to form a 5- or 6-membered carbocycle or a 5- or 6- membered heterocycle, each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CpQalky], Q^alkoxy.  and mono- or di-(CrC6alkyf)amino; U and T are independently N or CR7;
and mono- or di-(CrC6alkyf)amino; U and T are independently N or CR7;
R2 represents from 0 to 4 substituents independently chosen from Ci-C4alkyl, Cs-C^aloalkyl and oxo; or two R2 groups are taken together to form a methylene, ethylene, propylene or -CH2-O-CH2- bridge;
R3 is hydrogen, CrC4alkyl or Ci-Qhaloalkyl; R4 is:
(i) hydrogen; or
(ii) Ci-C3alkyl or C2-C8alkenyl, each of which is substituted with from O to 2 substituents independently chosen from hydroxy, amino, oxo, Q-Qalkoxy, Ci-Qalkanoyioxy, mono- or di-(Ci-QalkyI)amino, mono- or dKCj-CealkyOaminocarbonyl and (4- to 7-membered h eterocy c loalky l)C0-C2alky ! ;
Each R7 is independently hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyi, aminosulfonyl, Ci-C6alkyl, C2-Q,alkenyl, C2-C6alkynyl, CrC6alkoxy, CrQalkanoyl, C3- C6alkanone, C]-C6alkanoyloxy, C2-C6alkyl ether, C]-C6alkoxycarbonyl, C,-C6alkylthio, Cp C6alkylsulfonyl, mono- or di-(Ci-C6alkyl)aminoC0-C4alky], mono- or di-(Cr C6alkyI)aminocarbonylCo-C4alkyl, mono- or di-(Ci-C6alkyl)aminosulfonylCo-C4alkyl, C1- C6haloalkyl, Ci-C6haloalkoxy, CpQhydroxyalkyl, CpCe-uninoalkyl, C]-C6cyanoaikyl, or mono- or di-(Cj-C6alky!)aminoCo-C4alkyl;
R8 is hydrogen, CrCsalkyl, C2-C8alkenyl, C2-C8alkynyl, C2-C3alkyl ether, mono- or di-(Cr Csalkyl)aminoCo-C6alkyl, (C3-C7cycloalkyl)Co-C6alkyI or (5- to 10-membered heterocycle)Co-Cnalky], each of which is substituted with from O to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyi, imino, aminosulfonyi, -COOH, cyanoimido, Cj-C6alkyl, C,-C6alkoxy, Ci-C6haloalkyl, Cp Cfthaloalkoxy, mono- or di-(Ci-C6alkyi)amboC0-C6a]kyl, Ci-C6alkylsulfonyl, CrC6alkylthio, Ci-C6alkylaminosulfonyl, CrC6aikoxycarbonyl or Ci-C6alkanoylamino;
R9 and R15 are independently halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyi, aminosulfonyl, CrC6alky], C2-C6alkenyl, C2-C6aikynyl, Cj-Caalkoxy, Ci-C6alkanoyi, C3- C6a!kanone, CrC6a]kanoyloxy, C2-C6alkyl ether, Ci-C6alkoxycarbonyl, CrC6alkylthio, Cj- C6alkylsulfonyl, mono- or  mono- or di-(Cr C6alky!)aminocarbonylCo-C4alkyl, mono- or di-(C]-C6alkyl)aminosulfonyIC{)-C4alkyl, C|- C6ha!oaikyi, C]-C6haioalkoxy, Ci-C6hydroxyalkyl, C]-C6aminoalky], C,-C6cyanoalkyl, or mono- or di-(CrC6alkyl)aminoCo-C4alkyl;
mono- or di-(Cr C6alky!)aminocarbonylCo-C4alkyl, mono- or di-(C]-C6alkyl)aminosulfonyIC{)-C4alkyl, C|- C6ha!oaikyi, C]-C6haioalkoxy, Ci-C6hydroxyalkyl, C]-C6aminoalky], C,-C6cyanoalkyl, or mono- or di-(CrC6alkyl)aminoCo-C4alkyl;
Or R8 is taken together with R9 to form a fused heterocyc loalky I; and Rio is hydrogen, Q-Csalkyl, C2-C6alkenyl or C2-C6alkynyl.
2. A compound or salt or hydrate thereof according to claim 1, wherein the compound has the formula:

3. A compound or salt or hydrate thereof according to claim I , wherein the compound has the formula:

4. A compound or salt or hydrate thereof according to claim 1 , wherein the compound has the formula:

5. A compound or salt or hydrate thereof according to any one of claims 1-4, wherein Xs C(=O).
6. A compound or salt or hydrate thereof according to any one of claims 1-4, wherein X 
7. A compound or salt or hydrate thereof according to any one of claims 1 -4, wherein X is Cf=O)N(R11).
S. A compound or salt or hydrate thereof according to any one of claims 1-4, wherein X is CC=O)-[C(R1 ] Ri2)JvY.
9. A compound or salt or hydrate thereof according to any one of claims 1-8, wherein two R-2 groups are taken together to form a bridge.
10. A compound or salt or hydrate thereof according to claim 9, wherein the group
designated 
1 1. A compound or salt or hydrate thereof according to any one of claims 1- 10, wherein R2 represents from 0 to 4 substituents independently chosen from Ci-C4alkyl and Ci-Gihaloalkyl.
12. A compound or salt or hydrate thereof according to any one of claims 1-11, wherein RA is (C3-C7cycloalkyl)Co-C2alkyl.
13. A compound or salt or hydrate thereof according to ciaim 12, wherein the compound has the formula:

14. A compound or salt or hydrate thereof according to claim 13, wherein the compound has the formula:


15. A compound or salt or hydrate thereof according to any one of claims 1-1 1, wherein RA is 6- to 10-membered aryl or 5- to I O-membered heteroaiyl .
16. A compound or salt or hydrate thereof according to claim 15, wherein:

A and E are independently N or CR5; B, J and D are independently N or CR6; Each R5 is independently:
(i) hydrogen, halogen, nitro, cyano, or -COOH; or
(ii) CrQalkyi, C2-C6alkeπyl, C2-C6aikynyl, CrC6alkanoyi, C3-C6alkanone, C2-C6alkyl ether, C,-
C6alkoxycarbonyl, CrC6alkyisulfonyl or Cj-Cehaloalkyl; each of which is substituted with from 0 to 3 substituents independently chosen from hydroxy, amino, cyano, Cj-C6alkoxy and mono- or di-(CrC6alkyl)amino; or
(iii) taken together with a Rn or R12 moiety to form a C5-C7 eye loalkyl or a 5- to 7-membered heterocycloalkyl; and Each R6 is independently: (i) hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl; (ii) Ci-Qalkyl, C2-C6alkenyl, C2-C6alkynyl, CrC6alkoxy, Q-Qalkanoyl, CrC6aikyithio, Q- Cfialkanoyloxy, Ci-Cealkoxycarbonyi, Ci-Csalkylsulfonyl, Ci-Cehaloalkyl, mono- or di-(Cj- C6aIky])aminoCo-C4alkyI, mono- or di-(CrQalkyl)aminocarbonylCo-C4alkyI, mono- or di- (Ci-C6aIkyl)aminosulfonylCo-C4alkyl, (C3-C7cycloalkyi)C0-C2aIkyL (4- to 10-membered heterocycle)C0-C2alkyl, phenylCo-C2alkyl or (3- to 10-membered cycle)Co-C2aikoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C]-C4alkyl, C;-C4alkoxy, Ci-C4haloalkyl, C|-C4haloalkoxy, mono- or di-(Ci-C6aIkyl)amino, or 5- or 6-membered heterocycloalkyl; or two adjacent R^ groups are taken together to form a 5- or 6-membered ring that is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C]-C4alkoxy, C]-C4haloalkyl, and mono- or di-(Ci-C6aikyl)amino,
17. A compound or salt or hydrate thereof according to claim 16, wherein the compound has the formula:

18. A compound or salt or hydrate thereof according to claim 17, wherein the compound has the formula:

19. A compound or salt or hydrate thereof according to claim 16, wherein the compound has the formula:

20. A compound or salt or hydrate thereof according to claim 19, wherein the compound has the formula:

21. A compound or salt or hydrate thereof according to claim 16, wherein the compound has the formula:

22. A compound or salt or hydrate thereof according to claim 21, wherein the compound has the formula:


23. A compound or salt or hydrate thereof according to any one of claims 16-22, wherein at least one of B, J and D is substituted carbon.
24. A compound or salt or hydrate thereof according to claim 23, wherein I is substituted carbon.
25. A compound or salt or hydrate thereof according to claim 23 or ciaim 24, wherein B is substituted carbon.
26. A compound or salt or hydrate thereof according to claim 23, wherein two of B, J and D are carbon that is substituted with a substituent independently chosen from methyl, ethyl, halogen, C1-C2ha.oa.kyl and Ci-C2alkoxy.
27. A compound or salt or hydrate thereof according to any one of claims 16-26, wherein A, J and E are optionally substituted carbon and D is N.
28. A compound or salt or hydrate thereof according to any one of claims 16-27, wherein A and E are CH.
29. A compound or salt or hydrate thereof according to claim 12, wherein: 
A, B, E and D are independently N or CR6;
G is NR6. S or O; and
Each R5 is independently:
(i) hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl; (ii) CrC6alkyl, C2-C6atkenyL C2-C6alkynyl, C,-C6alkoxy, Ci-C6alkanoyl, CrC6alkylthio. C,- C6alkanoyloxy, Cj-C6alkoxycarboπyL Ci-C6alkylsulfonyl, CrCήhaloalkyl, mono- or di-(C3- C6alkyl)aminoC0-C4alkyl. mono- or di-(Ci-C6aIkyl)aminocarbonylC0-C4alkyL mono- or di- (Ci-C6alkyl)aminosulfonylCo-C4alkyl, (CrC7cycloalkyl)Co-C2alkyL (4- to 10-membered heterocycle)Co-C2alkyl, phenylC0-C2alkyl or (3- to 10-membered cycle)C0-C2alkoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C]-C4alkyl, Ci-C4alkoxy, Cj-Cjhaloalkyl, Ci-C4haloalkoxy, mono- or di-fCi-CealkylJamino. or 5- or 6-membered heterocycloalkyl; or
(iii) taken together with a Rn or R12 moiety to form a Cs-Cycycloaikyl or a 5- to 7-membered heterocycloalkyl; or two adjacent Rg groups are taken together to form a 5- or 6-membered ring that is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C]-C4alkyi. Ci-C4alkoxy, Ci-C4haloalkyl, and mono- or di-(Ci-C6alkyl)amino.
30. A compound or salt or hydrate thereof according to claim 29, wherein:

31. A compound or salt or hydrate thereof according to any one of claims 16-30, wherein each R6 is independently:
(i) hydrogen, halogen, hydroxy, nitro or cyano; or
(ii) C|-C6alkyl, C2-C6aikenyl, C2-C6alkynyl, CrC6alkoxy, C,-C6alkanoy!, Ci-C6alkylthio, C1- C6aJkanoyioxy, CrC6alkoxycarbonyl, Ci-Qalkylsulfonyl, C.-Cshaloalkyl. (Qr C7cycloalkyl)Co-Ciaikyl, (4- to 7-membered heterocycle)Co-C2alkyl or phenylC0-C2alkyl; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo,  CrC4alkoxy, Ci-C4haloalkyl, Cr C4haloalkoxy, mono- or di-(Cϊ-C6alkyl)amino and 5- or 6-membered heterocycloalkyl.
CrC4alkoxy, Ci-C4haloalkyl, Cr C4haloalkoxy, mono- or di-(Cϊ-C6alkyl)amino and 5- or 6-membered heterocycloalkyl.
32. A compound or salt or hydrate thereof according to any one of claims 1-31, wherein R3 is not H.
33. A compound or salt or hydrate thereof according to claim 32, wherein R;, is methyl
34. A compound or salt or hydrate thereof according to claim 33, wherein the group 
35. A compound or salt or hydrate thereof according to any one of claims 1 -34, wherein R8 is hydrogen, CrC6alkyl, C2-C6alkenyl. C2-C6alkynyl, mono- or di-(C]-C8alky!)aminoCi-C6alkyl or (5- to 7-membered heterocycle)C0-C(jalkyi, each of which is substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino. aminosulfonyl, Ci-C4alkyi, CrQalkoxy and mono- and di-(Cj-C8alkyJ)aminoCo-C6alkyl.
36. A compound or salt or hydrate thereof according to any one of claims 1-35, wherein R9 and R]5 are independently C]-C2alkyl, CrC2alkoxy or CpC2haioalkyl.
37. A compound of the formula:

X is C(=O), SO2, C(O)N(Rn), CC=O)-C(R11J=C(Rn), or C(=O)-[C(RπR!2)]v(Y)u; wherein Y is O or S; u is O or ] ; v is 1 or 2; and Rn and R]2 are independently H, Ci-C6alkyl, CrC6alkoxy, C2- C6alkyl ether, or taken together with a RB moiety to form a C5-C7cycloalkyl or a 5- to 7- membered heterocycloalkyi;
RA is 6- to 10-membered aryl, 5- to 10-membered heteroaryl, CrC6alkyl, CrC6haloalkyl, (C3- Cscycloalkyl)C0-C4alkyl or (5- to 7-membered heterocycIoaikyl)Co-C4alkyl, each of which is substituted with from O to 4 substituents independently chosen from RB; Each RB is independently:
(i) halogen, hydroxy, nitro, cyano. amino, oxo, -COOH, aminocarbonyl or aminosulfonyl;
(ii) CrQalkyi, CrCtalkeny!; Cr-Cealkynyi, C,-C6alkoxy; CrQaikanoyL Ci-C6alky!thio. C,-
Qalkanoyloxy, CrQalkoxycarbonyl, Cj-Cδalkyisulfonyl,  mono- or di-(Cj-
mono- or di-(Cj-
C6alkyl)aminoCo-C4aikyl, mono- or di-(CrCfialkyl)aminocarbonylC0-C4alkyl, mono- or di-
(CrC6alkyl)aminosulfonylCo-C4alkyl. (Ci-C7cycloalkyi)Co-C2alkyl, (4- to 10-membered heterocycle)Co-C2alkyl, phenylC0-C2alkyi or (3- to 10-membered cycle)C0-C2alkoxy; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C,-C4alkyi. Cj-C4alkoxy, Ci-Qhaloalkyl, CrC4haloalkoxy. mono- or di-(Ci-C6alkyl)amino and 5- or ό-membered heterocycloalkyi; or
(iii) taken together with a R1 1 or R12 moiety to form a C;-C7cycloalkyl or a 5- to 7-membered heterocycloalkyi; or two adjacent RB groups are taken together to form a 5- or 6-membered carbocycle or a 5- or 6- membered heterocycle, each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CrC4alkyl. Ci-C4alkoxy, C]-C4haloalkyl, and mono- or di-(Ci-C6alkyl)amino; U and T are independently N or CR7; P is N or CR15; Q is N or CR9; R] is cyan o, nitro, halogen or a group of the formula -L-M, wherein:
L is a single covalent bond, O, C(=O), OC(=O), C(=O)O, 0C(=O)0s S(O)W) N(Rx), C(O)N(Rx), N(RX)C(=O), N(RX)S(O)W, or S(O)WN(RX), wherein each Rx is independently hydrogen, Q- Qalkyl, C2-C6alkenyl, C2-C6alkynyl or Ci-C6haloaikyl and w is independently selected at each occurrence from 0, 1 and 2;
M is hydrogen, CrQalkyl, C2-C3alkenyl, C2-C3aikynyl, C2-C3alkyl ether, mono- or di-(Cr Csalkyl)aminoC0-C6alky!, (C3-C7cycloalkyl)Co-C6alky] or (5- to 10-membered heterocycle)C0-C6alkyl, each of which is substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosulfonyl. -COOH, cyanoimido,  Ci-Qhaloalkyl, CrC6alkoxy, Cr Cghaloalkoxy, Cϊ-C6alkylthio, C|-C6ailkylsu!fonyl, Ci-C6alkanoyl, CrC6alkanoyloxy, C|- C6alkanoylamino. Ci-C6alkylaminosulfonyl, Cj-Cealkoxycarbonyl, and mono- and di-(Cp C6alkyl)aminoC0-C6alkyl; Or R1 is taken together with R9 to form a fused C5-C8cycloalkyi or 5- to 8-membered heterocycloalkyl;
Ci-Qhaloalkyl, CrC6alkoxy, Cr Cghaloalkoxy, Cϊ-C6alkylthio, C|-C6ailkylsu!fonyl, Ci-C6alkanoyl, CrC6alkanoyloxy, C|- C6alkanoylamino. Ci-C6alkylaminosulfonyl, Cj-Cealkoxycarbonyl, and mono- and di-(Cp C6alkyl)aminoC0-C6alkyl; Or R1 is taken together with R9 to form a fused C5-C8cycloalkyi or 5- to 8-membered heterocycloalkyl;
R2 represents from 1 to 4 substituents independently chosen from C]-C4alkyl, C]-Cfhaloalkyl and oxo; R3 is hydrogen, C|-C4aikyl or CpGthaloalkyl; R4 is:
(i) hydrogen; or
(ii) C]-C8alkyl or C2-C8aikenyl, each of which is substituted with from 0 to 2 substituents independently chosen from hydroxy, amino, oxo, C|-C6alkoxy, CrC6alkanoyloxy, mono- or di-(CrC6alkyl)amino, mono- or di-(C|-C6alkyl)aminocarbonyI and (4- to 7-membered heterocycIoaikyI)Co-C2alkyl;
Each R7, R9 and RJ5 are independently hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl, aminosulfonyl, CpCβalkyl, C2-C6alkenyl, C2-C6aikynyl, CrC6alkoxy, C]- C6alkanoyl, C3-C6alkanone, CrCsalkanoyloxy, C2-C6alky] ether, CrC6alkoxycarbonyl, Cr Cήalkylthio, CrCόalkylsuIfonyl, mono- or di-(CrC6aikyl)ammoCo-C4aIkyL mono- or di-(Cr C6a!kyl)aminocarbonylCo-C4alky], mono- or di-(C]-C6aikyI)aminosuIfonylCo-C4alkyl, Ci- C6haloalkyl, Cj-C6haloalkoxy, Ci-C6hydroxyalkyl, Ci-C6aminoalkyl, Ci-C6cyanoalkyl, or mono- or di-(Ci-C6alkyl)aminoC0-C4aikyi; and Or R9 is taken together with R] to form a fused cycloalkyl or heterocycloalkyl.
38. A compound or salt or hydrate thereof according to claim 37, wherein the compound has the formula:

39. A compound or salt or hydrate thereof according to claim 37 or claim 38, wherein RA is 6- to 10-membered aryl or 5- to 10-membered heteroaiyl,
40. A compound or salt or hydrate thereof according to claim 39. wherein:

A and E are independently N or CR5; B, J and D are independently N or CR6; Each R5 is independently:
(i) hydrogen, halogen, nitro, cyano, amino, -COOH, aminocarbonyl or aminosuϊfonyl; or fii) Ci-Qalkyl, C2-Cήalkenyl, C2-C6alkynyi, CrC6aikanoyI, C3-C6alkanone, C2-C6alkyl ether, Cr C6alkoxycarbonyl, mono- or di-(C]-C6aikyl)amino, mono- or di-(Ci-C6alkyI)aminocarbonyl, mono- or di-(Ci-C6alkyl)aminosulfonyl, Ci-C6alkylsulfonyl or CrQhaloaikyl; each of which is substituted with from 0 to 3 substituents independently chosen from hydroxy, amino, cyano, Ci-C6a3koxy and mono- or di-(CrC6aikyl)amino; or (iii) taken together with a Rn or Ri2 moiety to form a C5-C7cycloalkyi or a 5- to 7-membered heterocycloalkyl; and Each R(, is independently:
(i) hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl; (ii) CrQalkyl. C2-C6alkenyl, C2-C6alkynyl, C]-C6alkoxy, C,-C6alkanoyl, CrC6alkylthio, C,- C6alkanoyloxy. CrC6alkoxycarbonyl, CrC6alkylsuIfonyl, Ci-C6haloalkyl, mono- or di-(C|- Cfialkyl)aminoCo-C4alkyl, mono- or di-(C|-C6alkyl)amiiiocarbonylCo-C4aikyl, mono- or di- (C]-C6alkyI)aminosulfonylCo-C4a]kyI, (C3-C7cyc]oalkyl)Co-C2alkyl, (4- to 10-membered heterocycle)Co-C2alkyl. pheny!C0-C2alkyl or (3- to 10-membered cycle)C0-C2aIkoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano. halogen, amino, oxo, C,-C4alkyL C]-C4alkoxy, CrC4haloalkyϊ, Cj-C4haloalkoxy, mono- or di-(CrQalkyl)amino, or 5- or 6-membered heterocycloalkyl; or two adjacent R6 groups are taken together to form a 5- or 6-membered ring that is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C]-C4alkoxy, Ci-C4haloalkyl, and mono- or di-(Ci-Cήalkyl)amino.
41. A compound or salt or hydrate thereof according to claim 40, wherein: each R5 is independently:
(i) hydrogen, halogen, nitro. cyano or -COOH; or
(ii) CVQalkyl, C2-C5alkerty], C2-C6alkynyl, CrC6alkanoyl, C3-C6aikanone, C2-C6alky! ether. C1- C6alkoxycarbonyi, C;-C6alkyisulfonyl or CrQhaloalkyi; each of which is substituted with from O to 3 substituents independently chosen from hydroxy, amino, cyano, CrQaikoxy and mono- or di-(CrC6a!kyi)amino; and each R6 is independently:
(i) hydrogen, halogen, hydroxy, nitro, cyano or-COOH; or
(Ii) C]-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, CrC6aikoxy, d-C6aϊkanoyl, Cj-Cgalkyllhio, Cr C6alkanoyloxy. Ci-C6alkoxycarbonyl, CpQalkylsulfonyl, C|-C6haioalkyl, (C3- C7cycloalkyl)C0-C2alky], (4- to 10-membered heterocycle)C0-C2aikyi or phenyICo-C2alkyI; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Ci-C4alkyl, CrC4alkoxy, Ci-C4haioalkyl, C1- Qhaloalkoxy, mono- or di-(Ci-C6a!kyI)amino and 5- or 6-membered heterocycloalkyl; or two adjacent R^ groups are taken together to form a fused, 5- or 6-membered ring.
42. A compound or salt or hydrate thereof according to claim 40 or claim 41, wherein at least one of B, J and D is substituted carbon.
43. A compound or salt or hydrate thereof according to claim 42, wherein J is substituted carbon.
44. A compound or salt or hydrate thereof according to ciaim 42 or claim 43. wherein B is substituted carbon.
45. A compound or salt or hydrate thereof according to claim 44, wherein two of B, J and D are carbon that is substituted with a substituent independently chosen from methyl, ethyl, halogen, C]-C2haloalkyl and Cj-C2alkoxy.
46. A compound or salt or hydrate thereof according to any one of claims 40-45, wherein A, J and E are optionally substituted carbon and D is N.
47. A compound or salt or hydrate thereof according to any one of claims 40-46, wherein A and E are CH.
48. A compound or salt or hydrate thereof according to claim 39, wherein:
RA is H E-AyG 5 ~H EG-'Af or *Λ D=E? ; A5 B, E and D are independently N or CR^; G is NR6, S or O; and Each R6 is independently:
(i) hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl;
(ii) CrC6alkyl, C2-C6alkenyl, C2-C6alkyny!, CrC6aikoxy, CrC6a1kanoyl, CrC6aIkyithio, C,-
C6alkanoyloxy, Ci-Qalkoxycarbonyl, Cj-Qalkylsulfonyl, Ci-Qhaloalkyl, mono- or di-(Cr
C6alkyl)aminoCo-C4alkyl, mono- or di-(C,-C6alkyl)aminocarbonylCo-C4alkyl, mono- or di- tCi-C(;alkyl)aminosulfonyICo-C4alkyl, (C3-C 7cycloalkyi)Co-C2alky], (4- to 10-membered heteiOcycle)Co-C2aikyl, pheπylC0-C2alkyl or (3- to 10-membered cycle)Co-C2aikoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano. halogen, amino, oxo, C]-C4alkyl,  Ci-C4haloalkyl, Ci-C4haloalkoxy, mono- or di-(C|-CβaikyI)amino, or 5- or 6-membered heterocycloalkyl ; or
Ci-C4haloalkyl, Ci-C4haloalkoxy, mono- or di-(C|-CβaikyI)amino, or 5- or 6-membered heterocycloalkyl ; or
(iii) taken together with a Rn or R12 moiety to form a Cs-Cycycloalkyl or a 5- to 7-membered heterocycloal kyl ; or two adjacent R6 groups are taken together to form a 5- or 6-membered ring that is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano. halogen, amino, oxo, CrC4aIkyl, Cj-Gjalkoxy, Ci-C4haloalkyl, and mono- or di-(Ci-C6alkyl)amino.
49. A compound or salt or hydrate thereof according to claim 37 or claim 38, wherein RA is C,-C6alkyl, CrC6haloalkyl, (C3-C8cycloalkyl)Co-C4alkyl or (5- to 7-membered heterocycloalkyl)C0-C4aIkyl. each of which is substituted with from O to 4 substituents independently chosen from:
(i) halogen, hydroxy, nitro or cyano; and
(ii) CrC6alkyl, Ci-Cealkenyl, C2-C6alkyny!, Ci-C6alkanoyl, CrC6alkylthio. CrC6aikanoyloxy. CrC6alkoxycarbonyi. C,-C6alkyisulfonyl, CrC6haloalkyl, (C3-C7cycloalkyl)Co-C2alkyϊ, (4- to 7-membered heterocycle)C0-C2aikyl or phenyiC0-C2alkyI; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Cp C4aikyl, C,-C4alkoxy, Ci-Cjhaloalkyl, Ci-C4haloalkoxy, mono- or di-(Ci-C6alkyl)amino and 5- or 6-membered heterocycloalkyl.
50. A compound or salt or hydrate thereof according to claim 49, wherein RA is optionally substituted (C3-C7cycloalkyl)C0-C2alkyl.
51. A compound or salt or hydrate thereof according to claim 37 or claim 38, wherein RA is a 9- or 10-membered heteroaryl that is substituted with from 0 to 4 substituents independently chosen from:
(i) halogen, hydroxy, nitro or cyano; and
(ii) Q-Qalkyl, C2-C6alkenyf, C2-C6alkynyl, Q-Qalkanoyl, Q-Qalkylthio, C,-C6alkanoyloxy, Q-Qalkoxycarbonyl, Q-Qalkylsulfonyl, C,-C6haloalkyϊ, (C3 -C7cyc loalky I)C0-C2 alky 1, (4- to 7-membered heterocycle)C0-C2alkyϊ or phenylCo-C2alkyl; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Q- C4alkyl, CrC4alkoxy, C!-C4haloalkyl, CrC4haloalkoxy, mono- or di-(Ci-C6alkyl)amino and 5- or 6-membered heterocycloalkyl.
52. A compound or salt or hydrate thereof according to any one of claims 37-51 , wherein at least one of P and Q is substituted carbon.
53. A compound or salt or hydrate thereof according to claim 52, wherein Q is substituted carbon.
54. A compound or salt or hydrate thereof according to claim 53, wherein P and Q are both substituted carbon.
55. A compound or salt or hydrate thereof according to claim 52, wherein R9 and R] 5 are independently hydrogen, Q-Qalkyi, Q-Qalkoxy or Q-Qhaloalkyl.
56. A compound or salt or hydrate thereof according to claim 55, wherein R9 and R] 5 are both methyl.
57. A compound or salt or hydrate thereof according to claim 52, wherein R9 and RJ5 are independently CrQalkyl and each R7 is independently hydrogen, C|-C2alkyl, CrC2alkoxy or Q- C2haloalkyl.
58. A compound or salt or hydrate thereof according to any one of claims 37-57, wherein
59. A compound or salt or hydrate thereof according to claim 58, wherein: R) is 0-R8; and
R8 is hydrogen, Q-Csalkyl, C2-C3alkenyl, C2-C3alkynyl, Q-C3alkyl ether, mono- or di-(Q- Csalkyi)aminoCo-CfialkyI, (C3-C7cycloalkyI)Co-C6alkyl or (5- to 10-membered heterocyc Ie)C0- Qaikyl, each of which is substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosulfonyL -COOH, cyanoimido, Q-C6alkyl, Q-Qalkoxy, Q-Cβhaloalkyl, Q-Qhaloalkoxy, mono- or di-(C,- C6alkyI)aminoC0-C6alkyI, Ci-C6alkylsιιlfonyl, Ci-Cβaikylthio, Ci-Ccalkylaminosulfonyl, Cr Cealkoxycarbonyl or Cj-Cgalkanoylamino.
60. A compound or salt or hydrate thereof according to claim 59, wherein Rg is hydrogen, C]-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, mono- or di-(Ci-C3alkyl)aminoCrC6aikyl or (5- to 7- membered heterocyc]e)C0-C6alkyl, each of which is substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano. oxo, aminocarbonyl, imino, aminosulfonyi, Q-QalkyL CrC4alkoxy and mono- and di-(Ci-C8alkyl)aminoCo-C6alkyl.
61. A compound or salt or hydrate thereof according to claim 58, wherein R, is methoxy.
62. A compound or salt or hydrate thereof according to any one of claims 37-61, wherein R3 is not H.
63. A compound or salt or hydrate thereof according to claim 62, wherein R3 is methyl.
64. A compound of the formula:

X is C(=O), SO2, C(=O)N(Rπ), C(=O)-C(R, ,J=C(R12), Or Ct=O)-[C(RnR12)Jv(Y)11; wherein Y is O or S; u is 0 or 1; v is 1 or 2; and Rn and R) 2 are independently H, CrC6alkyi, Cj-C6alkoxy, C2- C6alkyl ether, or taken together with a RB moiety to form a C5-C7cycloalkyl or 5- to 7-membered heterocycloalkyl;
RA is 6- to 10-membered aryl, 5- to 10-membered heteroaryl, Cj-CβalkyL Q-Qhaloaikyl, (C3- Cscycloalky])Co-C4alkyl or (5- to 7-membered heterocycloalkyl)C0-C4alkyl, each of which is substituted with from 0 to 4 substituents independently chosen from RB; Each RB is independently:
(i) halogen, hydroxy, nitro, cyano, amino, oxo, -COOH, aminocarbonyl or aminosulfonyi; (ii) Ci-C6alkyl, C2-C6alkenyl, CrC6alkynyl, CrC6aikoxy, CrC6aikanoyi, C,-C6aikylthio, Cr C6aikanoyloxy, Cj-Cgaikoxycarbonyl, Ci-CήaJkylsulfony!, Ci-Cghaloalkyl, mono- or di-(Cr C6alkyl)aminoCo-C4alkyl, mono- or d i-(C i -C6alkyl)aminocarbony! C0-C4 alkyl. mono- or di- (CrC6alkyl)aminosu!fonyICo-C4alky], (C3-C7cycloalkyl)Co-C2aikyl, (4- to 10-membered heterocycle)Co-C2alkyi, phenylCo-C2alkyl or (3- to 10-membered cycle)Co-C2aikoxy; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Q^alkyl, CrC4alkoxy, C]-C4haloalkyl, Ci-C4haloalkoxy, mono- or di-(Ci-C6alkyi)amino and 5- or 6-membered heterocycloalkyl; or (iii) taken together with a Rn or Ri2 moiety to form a C5-C7cycloalkyl or a 5- to 7-membered heterocycloalkyl; or two adjacent R8 groups are taken together to form a 5- or 6-membered carbocycle or a 5- or 6- membered heterocycle, each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CrC4alkyl, C]-C4alkoxy, CrC4haloalkyI, and mono- or di-(CrC6alkyI)amino; U and T are independently N or CR7; Q is N or CR9; P is N, CH or CR15; Ri is a group of the formula -L-M, wherein:
L is a single covalent bond, O, C(=O), OC(=O), C(O)O, 0C(=0)0, S(0)w, N(Rx), Cf=O)N(Rx), N(Rx)CC=O), N(RX)S(O)W, or S(O)WN(RX), wherein each Rx is independently hydrogen, C1- C6aikyl, Q-Csalkenyl, C2-C6alkynyl or CrCβhaloalkyl and w is independently selected at each occurrence from O, 1 and 2;
M is hydrogen, CrC8alkyl, C2-C3alkenyl, C2-C8alkynyl, Ci-Qalkyl ether, mono- or di-(Cr Csalkyl)aminoC0-C6alkyi, (C3-C7cycloalkyl)Co-C6alkyl or (5- to 10-membered heterocycle)Co-C6alkyl, each of which is substituted with from O to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosulfonyl, -COOH, cyanoimido, Ci-Qalkyl, CrC6haloalkyl, CrC6alkoxy, C]- C6haloalkoxy, Ci-C6alkylthio, Ci-C6alkylsulfonyl, CpQalkanoyl, Ci-Cgalkanoyloxy, Q- Qalkanoylamino, Ci-Cβalkylaminosulfonyϊ, Cj-Cfialkoxycarbonyl, and mono- and di-(C]- C6alkyI)aminoC0-C6alkyl; or Ri is taken together with R9 to form a fused C5-C8cycloalkyl or 5- to 8-membered heterocycloalkyl; such that R] is not hydrogen;
R2 represents from 1 to 4 substituents independently chosen from Ci-C4alkyl, Ci-C4haloalkyl and oxo; or two R2 groups are taken together to form a methylene, ethylene, propylene or -CH2-O-CH2- bridge;
R3 is hydrogen, CrC4aikyl or CrC4haloaIkyl; R4 is:
(i) hydrogen; or
(ii) C]-C3alkyl or C2-Cgalkenyl, each of which is substituted with from O to 2 substituents independently chosen from hydroxy, amino, oxo, Ci-C6alkoxy, CrC6aikanoyloxy, mono- or di-(Ci-C6alkyI)amino, mono- or di-(CrC6aikyl)aminocarbonyl and (4- to 7-membered heterocy cloalky l)Co-C2alky 1 ; Each R7 is independently hydrogen, halogen, hydroxy, nitro, cyano, amino. -COOH. aminocarbonyL aminosulfonyl, Ci-Cgalkyl, C2-C6alkenyl, C2-C6alkynyl, Cj-Qalkoxy, Ci-Qaikanoyl, C3- Cftalkanone, Q-Qalkanoyloxy, C2-C6a]kyl ether, Ct -C6alkoxycarbonyl, CrC6aikyIthio, Cr C6aikylsulfonyL mono- or di-(C]-C6alkyl)aminoC0-C4a]ky!, mono- or di-(Cr C6alkyI)aminocarbonylC0-C4alkyl, mono- or di-(Cs-C6alkyl)aminosιtIfonylCo-C4alkyl, Q- C6haloalkyl, CrC6haloalkoxy, CrQhydroxyalkyI, CrC6aminoalkyl, Ci-Cgcyanoalkyl, or mono- or di-(C]-C6aikyl)aminoCo-C4aikyI; and
R9 and Rj5 are independently halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyi, aminosulfonyi, CrC6alkyI, C2-Q,alkenyl, C2-C6alkynyl, CrCβaikanoyl, CrC6alkanone, C|- Qaikanoyloxy,  ether, CrC6alkoxycarbonyI, C]-C6alkylthio, C]-C6alky!suifonyl, mono- or di-(Ci-C6aIkyl)aminoCo-C4alkyl, mono- or di-(Ci-C6alkyl)aminocarbonylCo-C4alkyl, mono- or di-(CrC6alkyl)aminosulfonylCo-C4alkyl, CrCβhaloaikyl, Ci-C6haloalkoxy, C]- C6hydroxyalkyl, CrCeaminoalkyl, CrC6cyanoaikyI, or mono- or di-(Ci-C6alkyl)aminoC0- C4alkyl;
ether, CrC6alkoxycarbonyI, C]-C6alkylthio, C]-C6alky!suifonyl, mono- or di-(Ci-C6aIkyl)aminoCo-C4alkyl, mono- or di-(Ci-C6alkyl)aminocarbonylCo-C4alkyl, mono- or di-(CrC6alkyl)aminosulfonylCo-C4alkyl, CrCβhaloaikyl, Ci-C6haloalkoxy, C]- C6hydroxyalkyl, CrCeaminoalkyl, CrC6cyanoaikyI, or mono- or di-(Ci-C6alkyl)aminoC0- C4alkyl;
Or R9 is taken together with Rj to form a fused cycloalkyl or heterocycloalkyl; and
Rio is hydrogen, Ci-C&a1kyl, C2-C6alkenyl or C2-C6alkynyl.
65. A compound or salt or hydrate thereof according to claim 64, wherein n is O and m is I, 2 or 3.
66. A compound or salt or hydrate thereof according to claim 64, wherein n is 1 and m is 2 or 3.
67. A compound or salt or hydrate thereof according to claim 64, wherein n is 2 and m is O, 3 or 2.
68. A compound or salt or hydrate thereof according to claim 64, wherein n is 3 and m is O or 1.
69. A compound or salt or hydrate thereof according to claim 64, wherein the compound has the formula:

70. A compound or salt or hydrate thereof according to claim 64, wherein the compound has the formula:

71. A compound or salt or hydrate thereof according to any one of claims 64-70, wherein RA is 6- to 10-membered aiyl or 5- to 10-membered heteroaryl.
72. A compound or salt or hydrate thereof according to claim 71 , wherein:

(i) hydrogen, halogen, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyi; or (ii) CrC6alky], C2-C6alkenyl, CrC6alkynyl, C,-C6alkanoy], C3-C6alkanone, CrC6alky! ether, C1- Cftalkoxycarbonyl. mono- or di-(CrC6alkyl)amino, mono- or di-(Ci-C6alkyl)amiπocarbony!, mono- or di-(Ci-Q,alkyi)aminosulfonyl, Ci-Cβalkylsulfonyl or CrC6haloa!kyl; each of which is substituted with from 0 to 3 substituents independently chosen from hydroxy, amino, cyano, d-C6alkoxy and mono- or di-(CrC6alky])amino; or (iii) taken together with a Rn or Ri 2 moiety to form a C5-C7cycloalkyl or a 5- to 7-membered heterocycloalkyl; and Each R6 is independently:
(i) hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosuifonyl; (ii) CrQalkyl, C2-C6alkenyL C2-C3alkynyL C,-C6alkoxy, C,-C6alkanoyl, C,-C6alkylthio, C,- Qalkanoyloxy, Ci-C6alkoxycarbonyl, C,-C6alkylsulfonyl, CrC6haloalkyl, mono- or di-(Cr C6alkyl)aminoC0-C4aikyl, mono- or di-(Ci-C6alkyl)aminocarbonylCo-C4aϊkyls mono- or di- (Ci-C6alkyl)aminosuIfonylCo-C4alkyl, (C3-C7cycloalkyl)C0-C2aikyl, (4- to 10-membered heterocyc1e)Co-C2aIkyl, ρhenylC0-C2alkyi or (3- to 10-membered cycle)C0-C2alkoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Quality!,  CrC4haloalkyl, Ci-C4haloalkoxy, mono- or di-(C]-C6aIkyl)amino, or 5- or 6-membered heterocycloaikyl; or two adjacent R6 groups are taken together to form a 5- or 6-membered ring that is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C]-C4alkyl, Ci-C4alkoxy, Ci-C4haloalkyi, and mono- or di-(Ci-Qalkyl)amino.
CrC4haloalkyl, Ci-C4haloalkoxy, mono- or di-(C]-C6aIkyl)amino, or 5- or 6-membered heterocycloaikyl; or two adjacent R6 groups are taken together to form a 5- or 6-membered ring that is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C]-C4alkyl, Ci-C4alkoxy, Ci-C4haloalkyi, and mono- or di-(Ci-Qalkyl)amino.
73. A compound or salt or hydrate thereof according to claim 71 or claim 72, wherein: each R5 is independently:
(i) hydrogen, halogen, nitro, cyano or --C00H; or
(ii) CrQ,alkyl, C2-C6alkenyl, C2-C6alkynyL CrC6alkanoyl, C3-C6alkanone, C2-C6alkyl ether, Cr C6alkoxycarbonyl, C]-C6alkyisulfonyl or C]-C6haloalkyl; each of which is substituted with from 0 to 3 substituents independently chosen from hydroxy, amino, cyano, Q-Qaikoxy and mono- or di-(Cj-C6alky])amino; and each Rβ is independently:
(i) hydrogen, halogen, hydroxy, nitro, cyano or-COOH; or
(ii) CrQalkyl, C2-C6alkenyl, C2-C6alkynyl, CrC6alkoxy, CrC6alkanoyl, CrC6alkylthio, Cr C6alkanoyloxy, CrQaϊkoxycarbonyl, CrC6alkylsulfoπyl, C|-C6haloalkyl. (C3- C7cycloaikyl)Co-C2alkyl, (4- to 7-membered heteiOcycle)Co-C2alkyl or phenylCo-C2alkyl; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CpQalkyl, Ci-C4alkoxy, C]-C4haloalkyl, C1- C4haloalkoxy, mono- or di-(CrQalkyl)amino and 5- or 6-membered heterocycloalkyl; or two adjacent R6 groups are taken together to form a fused, 5- or 6-membered ring.
74. A compound or salt or hydrate thereof according to claim 72 or claim 73 wherein at least one of B, J and D is substituted carbon.
75. A compound or salt or hydrate thereof according to claim 74, wherein J is substituted carbon.
76. A compound or salt or hydrate thereof according to claim 74 or claim 75, wherein B is substituted carbon.
77. A compound or salt or hydrate thereof according to claim 74. wherein two of B. J and D are carbon that is substituted with a substituent independently chosen from methyl, ethyl, halogen, CrC2haloalkyl and C,-C2alkoxy.
78. A compound or salt or hydrate thereof according to any one of claims 72-77, wherein A, J and E are optionally substituted carbon and D is N.
79. A compound or salt or hydrate thereof according to any one of claims 72-78, wherein A and E are CH.
80. A compound or salt or hydrate thereof according to claim 71 , wherein: 
A, B, E and D are independently N or CRe; G is NR6, S or O; and Each R6 is independently;
(i) hydrogen, halogen, hydroxy, nitro. cyano, amino, -COOH, aminocarbonyl or aminosulfonyl;
(ii) Ci-C6alky], C2-C6alkenyl, C2-C6alkynyl, CrC6aIkoxy, CrC6alkanoyl, CrC6alkyIthio, C1-
C6alkanoyloxy, C,-C6alkoxycarbonyϊ, C]-C6alkylsu]fonyl, CrQhaloalkyl, mono- or di-(Cr
C6alkyI)aminoCo-C4alkyI, mono- or di-(Ci-C6alkyl)aminocarboiiylC0-C4alkyi, mono- or di-
(C!-C6alkyl)aminosulfonylCo-C4alkyl, (C3-C7cycloaIky!)Co-C2aIky!, (4- to 10-membered heterocycle)Co-C2alkyl, ρheny]C0-C3alkyl or (3- to 10-membered cycle)C0-C2a!koxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CrC4a!kyi, CrC4alkoxy, Ci-C4haloalkyl, CrCjhaloalkoxy, mono- or di-(Cj-C6alkyl)amino. or 5- or 6-membered heterocycloalkyi; or
(iii) taken together with a Rn or R12 moiety to form a C5-C7cycloalkyl or a 5- to 7-membered heterocycloalkyi; or two adjacent R6 groups are taken together to form a 5- or 6-membered ring that is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C,-C4aikyl, C|-C4alkoxy, C1-C4ha.oa.kyl, and mono- or di-(CrC6alkyl)amino.
81. A compound or salt or hydrate thereof according to any one of claims 64-70, wherein RA is CrQalkyl, CrC6haloaikyl, (C3-Cgcycloalkyi)C0-C4alky] or (5- to 7-membered heterocycloalkyl)Co-C4aJkyl, each of which is substituted with from 0 to 4 substituents independently chosen from:
(i) halogen, hydroxy, nitro or cyano; and
(ii) CrC6alkyl, C2-C6aikenyl, C2-C6alkynyi, CrC6aikanoyl, CrC6aikylthio, CrC6alkanoyloxy, C,-C6alkoxycarbonyl, CrC6alkylsu]fonyl, C,-C6haloalkyL (C3-C7cycloalkyl)Co-C2a!kyl, (4- to 7-mesτsbered heterocycle)C0-C2alkyl or phenylC0-C2alkyl; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Cr C4alkyl, C|-C4alkσxy, CrC4haloalkyl! CrC4haloalkoxy, mono- or di-(CrC6alkyl)amino and 5- or 6-membered heterocycioalkyl.
82. A compound or salt or hydrate thereof according to claim 81, wherein RA is optionally substituted 
83. A compound or salt or hydrate thereof according to any one of claims 64-82, wherein at least one of P and Q is substituted carbon.
84. A compound or salt or hydrate thereof according to claim 83, wherein Q is substituted carbon .
85. A compound or salt or hydrate thereof according to claim 84, wherein P and Q are both substituted carbon.
86. A compound or salt or hydrate thereof according to claim 83, wherein R9 and R45 are independently CrC2alkyl, CrC2aikoxy or Ci-C2haioalkyl.
87. A compound or salt or hydrate thereof according to claim 86, wherein R9 and R]5 are each methyi.
88. A compound or salt or hydrate thereof according to claim 83, wherein R9 and R! 5 are independently chosen from CrC4alkyl and each R7 is independently hydrogen, CrC2alkyl, C1- C2alkoxy or CrC2haloalkyI.
89. A compound or salt or hydrate thereof according to any one of claims 64-88, wherein: 
Rg is hydrogen, C]-C8alkyl, C2-C3a!kenyl, C2-C8alkynyl, C2-C3a]kyl ether, mono- or di-(Cr C8alkyl)aminoCo-C6alkyl, (Cs-Cvcycloalky^Co-Cealkyl or (5- to 10-membered heterocycle)C0- C6alkyl, each of which is substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosuifonyl, -COOH, cyano imido, Q-Qalkyl, Ci-C<-,alkoxy, Ci-Cβhaloalkyl, Ci-C6haloaikoxy, mono- or di-(C,- C6aikyl)aminoCo-C6alkyI, d-Cgalkylsulfonyl, CrC6alkyithio, CrC6aIkylaminosulfonyl, Cr Cβalkoxycarbonyl or CrQalkanoylamino.
90. A compound or salt or hydrate thereof according to claim 89, wherein Rs is hydrogen, C-Qalkyl, C2-C6aikenyl, C2-C6alkynyl, mono- or di-(C]-C8alky!)aminoCi-C6alkyl or (5- to 7- membered heterocycIe)C0-C6aikyl, each of which is substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosuifonyl, CrC4alkyl, CrC4alkoxy and mono- and di-(Ci-C8alkyl)aminoC0-C6alkyl.
91. A compound or salt or hydrate thereof according to claim 89, wherein Rj is methoxy.
92. A compound or salt or hydrate thereof according to any one of claims 64-91, wherein R3 is not H.
93. A compound or salt or hydrate thereof according to claim 92, wherein R3 is methyl.
94. A compound or salt or hydrate thereof according to any one of claims 64-93, wherein two R2 groups are taken together to form a bridge.
95. A compound of the formula:

RA is 6- to 10-membered aryl, 5- to 10-membered heteroaryl, Cj-C6a!kyl, Ci-Cehaioalkyl, (C3- Cscycloa]kyi)Co-C4alky! or (5- to 7-membered heterocycϊoaikyl)C0-C4alkyI, each of which is substituted with from 0 to 4 substituents independently chosen from RB; Each RB is independently:
(i) halogen, hydroxy, nitro, cyano, amino, oxo, -COOH, aminocarbonyl or aminosuifonyl; or (ii) Ci-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, CrC6alkoxy, C|-C6alkanoyl. C]-C6alkyithio, Cr C6alkanoyloxy, CrC6alkoxycarbonyl, C]-C6alkylsulfonyl, CrQhaloalkyl, mono- or di-(Cr C6alkyl)aminoCo-C4aIkyl, mono- or di-(Ci-C6alkyl)aminocarbonylCo-C4alkyl, mono- or di- (CrC6alkyl)aminosulfonylC0-C4aikyl, (C3-C7cycloalkyl)Co-C2alkylJ (4- to 10-membered heterocycle)Co-C2alkyl, phenylCo-Cjalkyl or (3- to 10-membered cycle)Co-C2alkoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C,-C4alkyl, C|-C4alkoxy, C|-C4haioaikyl, Ci-C4haloalkoxy, mono- or di-(C]-C6alky!)amiπo, or 5- or 6-membered heterocycloalkyl; or two adjacent RB groups are taken together to form a 5- or 6-membered carbocycle or a 5- or 6- membered heterocycle, each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Ci-C4alkyl, Q-Qalkoxy, C|-C4haioalkyi, and mono- or di-(Cj-C6alkyl)amino; U and T are independently N or CR/?; Ri is cyano, nitro. halogen or a group of the formula -L-M, wherein:
L is a single covalent bond, O, C(O), 0C(=0), C(=O)O, OC(O)O, S(0)w, N(R11), C(O)N(Rx), N(Rx)C(O). N(RX)S(O)W, or S(O)WN(RX), wherein each Rx is independently hydrogen. C1- Qalkyi, C2-C6alkeny], C2-C6alkynyl or CpCehaloalkyI and w is independently selected at each occurrence from O, 1 and 2;
M is hydrogen. C(-C3aikyl, C2-C8alkenyl, C2-C3alkynyl. C2-C8alkyl ether, mono- or di-(Cr Csalkyl)aminoCo-C6alkyi. (C3-C7cycloalkyI)C0-C6alkyl or (5- to 10-membered heterocycle)Co-Qalkyi, each of which is substituted with from O to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo. aminocarbonyl, imino, aminosuifonyl, -COOH, cyanoimido, Q -Chalky!, Cj-Cghaloalkyl, Cj-C6alkoxy, C1- C6haloalkoxy, Ci-C6alkylthio, CrC6aIkylsulfonyl. CrC6alkanoyl, CrC6alkanoyloxy. Cr Qalkanoylamino, Q-Qalkylaminosulfonyl, Q-Qalkoxycarbonyl, and mono- and di-(Q- QalkyOaminoCo-Qaikyl; Or Rj is taken together with R9 to form a fused Q-Qcycloalkyl or 5- to 8-membered heterocycloalkyl;
R2 represents from 0 to 4 substituents independently chosen from CrC4alkyl, Ci-C4haloalkyl and oxo; or two R2 groups are taken together to form a methylene, ethylene, propylene or -CH2-O-CH2- bridge; R3 is hydrogen, Q-Qalkyl or CrC4haioaikyl; and
Each R7, R9 and R!5 are independently hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl, aminosuϊfonyl, Ci-C6alkyi, C2-QaI kenyl, Q-Qalkynyϊ. Q-Qalkoxy, Q- Qalkanoyl, Q-Qalkanone, Q-C6alkanoyloxy, C2-Qalkyl ether, Q-Qalkoxycarbonyl, Q- C6aikylthio, Q-Qsaikylsulfonyl, mono- or di-(Ci-C6alkyl)aminoCo-C4alkyl, mono- or di-(Q- C6alkyl)aminocarbonylCo-C4alkyl, mono- or di-CQ-QalkyOaminosulfonylCo-Qialkyl, Q- Qhaloalkyl, Q -Qhaloalkoxy, C]-C6hydroxyalky3, C]-C6aminoalkyl, CrC6cyanoalkyl, or mono- or di-(CrC6alky!)aminoCo-C4aikyl; or R9 is taken together with R1 to form a fused cycloalkyi or heterocycloalkyl.
96. A compound or salt or hydrate thereof according to claim 95, wherein RA is 6- to 30- membered aryl or 5- to 10-membered heteroaryl, each of which is substituted with from 0 to 4 substituents independently chosen from RB.
97. A compound or salt or hydrate thereof according to claim 96, wherein:

A and E are independently N or CR5; B, J and D are independently N or CR6; Each Rj is independently:
(i) hydrogen, halogen, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl; or (ii) CrC6alkyl, C2-C6alkenyi, C2-C6alkynyL CrC6aikanoyl, C3-C6alkanone, C2-C6aikyl ether, Q- Qalkoxycarbonyl, mono- or di-(Q-Qalkyl)amino, mono- or di-(Ci-C6alkyl)aminocarbonyl. mono- or dHQ-Qaiky^aminosulfonyi, C]-C6alkylsulfonyl or Q-Qhaloalkyi; each of which is substituted with from O to 3 substituents independently chosen from hydroxy, amino, cyano, Q-Qalkoxy and mono- or di-(Ci-C6alkyl)amino; or (iii) taken together with a Rn or R,2 moiety to form a Q-Qcycloalkyl or a 5- to 7-membered heterocycloalkyl; and Each R6 is independently: (i) hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl: (ii) CrC6alkyL C2-Qaikenyl, C2-C6alkynyl, C,-C6alkoxy, CrC6alkanoyl, C,-C6alkylthio, C1- C6alkanoyioxy, Ct-C6alkoxycarbonyl, CrC6alkylsulfonyl, Ci-C6ha]oalkyl, mono- or di-(Q- C6alkyl)aminoCo-C4alkyl, mono- or di-(Ci-C6alkyl)aminocarbonylCo-C4aikyl, mono- or di- (CrC(ialkyl)aminosulfonylCo-C^alkyL (C3-C7cycloalkyl)Co-C2aikyl, (4- to IO-membered heterocycIe)Co-C2aIkyl, phenyiC<rC2aikyl or (3- to 10-membered cycle)C0-C2alkoxy; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Ci-C4alky!, CrC4alkoxy, CrGihaloalkyI, Q-Qhaloalkoxy, mono- or di~(CrC6alkyl)amino, or 5- or 6-membered heterocycloaikyl; or two adjacent R$ groups are taken together to form a 5- or 6-membered ring that is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C]~C4alkyL Ci-C4alkoxy, CrC4haioaIkyl, and mono- or di-(C|-Cήalkyl)amino.
98. A compound or salt or hydrate thereof according to claim 97, wherein: each R5 is independently:
(i) hydrogen, halogen, nitro, cyano or ^COOH; or
(it) CpQalkyi, C2-C6alkenyl, CrC6alkynyl, CrC6alkanoyl, C3-C6alkanone, C2-C6alkyl ether, C1- Cealkoxycarbonyi, Cj-Cβalkylsulfonyl or CrC6haloalkyϊ; each of which is substituted with from O to 3 substituents independently chosen from hydroxy, amino, cyano, CrC6alkoxy and mono- or di-(C!-C<;aikyl)amino; and each R0 is independently:
(i) hydrogen, halogen, hydroxy, nitro, cyano or-COOH; or
(ii) C]-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, CrC6aikoxy, C,-C6alkanoyi, C,-C6aikylthio, C1- Cήalkanoyloxy, Ci-Cgalkoxycarbonyl, C]-C6alkylsuifonyi, CpQhaloaikyl, (C3- C7cycloalkyl)C0-C2aikyl, (4- to 10~membered heterocyc!e)Co-C2aikyl or pheny!C0-C2alkyl; each of which is substituted with from O to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, CrC4alkyl, CrC4aikoxy, C|-C4haloalkyi, C,- Cjhaloalkoxy, mono- or di-(C]-C6aikyl)amino and 5- or 6-membered heterocycloaikyl; or two adjacent R6 groups are taken together to form a fused, 5- or 6-membered ring.
99. A compound or sait or hydrate thereof according to claim 97 or claim 98, wherein at least one of B, J and D is substituted carbon.
100. A compound or salt or hydrate thereof according to claim 99, wherein J is substituted carbon.
101. A compound or sait or hydrate thereof according to claim 99 or claim 100, wherein B is substituted carbon.
102. A compound or salt or hydrate thereof according to claim 99, wherein two of B. J and D are carbon that is substituted with a siibstituent independently chosen from methyl, ethyl, halogen, Ci-Cahaloaikyl and Ci-C2alkoxy.
103. A compound or salt or hydrate thereof according to any one of claims 97-102, wherein A, J and E are optionally substituted carbon and D is N.
104. A compound or salt or hydrate thereof according to any one of claims 97-103, wherein A and E are CH.
105. A compound or salt or hydrate thereof according to claim 97, wherein RA is Cr C6alkyl, Cj-C6haloalkyl, (C3-C3cycloalkyl)C0-C4alkyl or (5- to 7-membered heterocycioalky!)C0- C4alkyi, each of which is substituted with from 0 to 4 substituents independently chosen from:
(i) halogen, hydroxy, nitro or cyano; and
(ii) Ci-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C,-C6alkanoyl, C,-C6alkylthio, C,-C6alkanoyioxy, C,-C6alkoxycarbonyl, CrQalkyIsulfonyl, C,-C6haloalky], (C3-C7cycIoalkyl)Co-C2aIkyl, (4- to 7-membered heterocycle)C0-C2alkyl or phenylC0-C2aikyl; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Cr C4alkyl, Q-^alkoxy, C]-C4haloalkyl, Q-C^aloalkoxy, mono- or di-(CrC6alkyl)amino and 5- or 6-membered heterocycloalkyl.
106. A compound or salt or hydrate thereof according to claim 105, wherein RA is optionally substituted (C3-C7cycioaikyl)C<rC2alkyi.
107. A compound or salt or hydrate thereof according to any one of claims 95-106, wherein at least one Of R9 and R]5 is not H.
108. A compound or salt or hydrate thereof according to claim 107, wherein R9 and Ri5 are both other than hydrogen.
109. A compound or salt or hydrate thereof according to claim ] 08, wherein R9 and R!5 are independently Ci-C2alkoxy or C|-C2haϊoaikyl.
1 10. A compound or salt or hydrate thereof according to claim 109, wherein R9 and Rj5 are each methyl
11 1. A compound or salt or hydrate thereof according to claim 109, wherein R9 and R15 are independently chosen from C,-C2alkyl and each R7 is independently hydrogen, CrC2alkyl, C)- C2alko>cy or CrC2haloaIkyl.
1 12. A compound or salt or hydrate thereof according to any one of claims 95-1 1 1, wherein Ri is not H.
1 13. A compound or salt or hydrate thereof according to claim 112, wherein: R, is O-Rs; and
Rs is hydrogen, CrCsalkyl, C2-Qa]kenyl, Ci-Csalkynyl, C2-C3a]kyl ether, mono- or di-(Cr Csa!kyl)aminoC0-C6alkyl, (Cj-CvcycIoalkyOCo-Cgalkyl or (5- to l ϋ-membered heterocycie)C0- Qalky], each of which is substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosulfonyl, -COOH, cyanoimido, C;-C6alky!, Q-Cealkoxy, Ci-C6haloalkyl, Ci-Cβhaloalkoxy, mono- or di-(C,- C6aIkyl)ammoC0-Qalkyl, Ci-C6a!kylsu!fonyl, Cj-C6a!ky]thio, Ci-Cealkylaminosulfonyl, Cr Qalkoxycarbonyi or Ci-Cόalkanoylamino.
1 14. A compound or salt or hydrate thereof according to claim 1 13, wherein Rg is hydrogen, Q-Qsaikyl, C2-C6alkenyl, C2-C6alkynyl, mono- or di-(C1-C8alkyl)aminoCi-C6alkyl or (5- to 7-membered heterocycle)Co-Qalkyi, each of which is substituted with from 0 to 3 substitυents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosulfonyl, Ci-C4alkyi, CrQalkoxy and mono- and di-(Ci-Csalkyi)aminoC0-C6alkyl.
1 15. A compound or salt or hydrate thereof according to claim 1 13, wherein Ri is methoxy,
1 16. A compound or salt or hydrate thereof according to any one of claims 95-115, wherein R3 is not H.
1 17. A compound or salt or hydrate thereof according to claim 1 16, wherein R3 is methyl,
1 18. A compound of the formula:
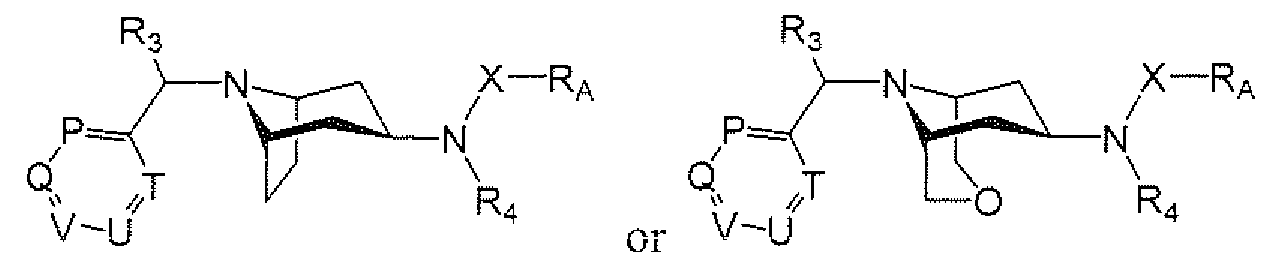
X is C(=O), SO2, Ct=O)N(Rn), C(=O)-C(Rπ)=C(R12)5 or C(O)-[C(R, ,R12XK(Y),; wherein Y is O or
S; u is O or 1 ; v is I or 2; and Rn and R12 are independently H, Cj-Qalkyl, CpQalkoxy, C2-
Cfialkyl ether, or taken together with a RB moiety to form a C5-C7cycloalkyl or a 5- to 7- membered heterocycloalkyl; RA is 6- to 10-membered aryl, 5- to 10-membered heteroaryl, Cj-Cgalkyl, Ci-C6haloafkyl, (C3-
CscycloaIkyl)Co-C4alkyl or (5- to 7-membered heterocycloalkyl)Co-C4alkyl, each of which is substituted with from O to 4 substituents independently chosen from RB; Each RB is independently:
(i) ha!ogen, hydroxy, nitro, cyano, amino, -COOH, oxo, aminocarbonyl or aminosulfonyl;
(ii) Ci-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, CrC6alkoxy, CrC6alkanoyl, C,-C6a!kylthio, Cr
Qalkanoyloxy, CrC6alkoxycarbonyI, CrC6alkyisulfonyI, CrC6haloalkyL mono- or di-(Cr
C6aIkyl)aminoCo-C4alkyls mono- or di-(C]-QalkyI)aminocarbonylCo-C4aikyI, mono- or di-
(C1-C6alkyl)aminosulfonylCo-C4alkyl, (C3-C7cycloalkyl)C<rC2a!kyl, (4- to 10-membered heterocycle)C0-C2alkyI, phenylC0-C2alky! or (3- to 10-membered cycle)C0-C2alkoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Ci -Chalky 1, Ci-C4alkoxy, C|-C4haloalkyl, Q-Qhaioalkoxy, mono- or di-(Ci-C6alky!)amino and 5- or 6-membered heterocycloalkyi; or
(iii) taken together with a Rn or R,2 moiety to form a C5-C7cycloalkyl or a 5- to 7-membered heterocyc 1 oalky 1; or two adjacent RB groups are taken together to form a 5- or 6-membered carbocycle or a 5- or 6- membered heterocycle, each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, Ci-C4alkyl, CrC4alkoxy, C,-C4haloalkyl, and mono- or di-(CrC6a]kyl)amino; V is N or CR1;
U and T are independently N or CR7; P is N or CR]5; Q is N or CR9; R1 is cyano, nitro. halogen or a group of the formula -L-M, wherein:
L is a single covalent bond, O, C(=0), OC(O)5 C(=0)0, OC(O)O, S(0)w, N(Rx), C(O)N(Rx), N(Rx)Cf=O), N(Rx)S(OXv, or S(O)WN(RS), wherein each Rx is independently hydrogen, C,- Qalkyl, C2-Qa! kenyl, C2-C6alkynyi or CrC6haloalkyl and w is independently selected at each occurrence from O, 1 and 2; and
M is hydrogen, CrC8alkyl, C2-C3alkenyl, CrCsa!kynyl, Ci-Csalkyl ether, mono- or di-(Cr Csalkyl)aminoCo-C6aikyl, (C3-C7cycloalkyl)C0-C6alkyl or (5- to 10-membered heterocyc Ie)C0-QaI kyl, each of which is substituted with from O to 3 substituents independently chosen from halogen, amino, hydroxy, cyano. oxo, aminocarbonyl, imino, aminosulfonyl, -COOH, cyanoimido, CrC6alkyl, CrC6haloalkyi, CrC6alkoxy, C,- C(,haloaikoxy, Ci-C6alkylthio, C]-C6alkyisulfonyl, CrC6alkanoyl, CrC6alkanoyIoxy, C,- C6alkanoylamino, Ci-Cealkylaminosulfonyl, CrC6a!koxycarbonyl, and mono- and di-(Cr CealkylJaminoCo-Cgaikyl; or Ri is taken together with R9 to form a fused C5-C3cycloalkyl or 5- to 8-membered heterocycloalkyi ;
R3 is hydrogen, C,-C4alkyl or CrC4haloaIkyl; R4 is: (i) hydrogen; or
(ii) CrCgalkyl or C2-Csalkenyl, each of which is substituted with from 0 to 2 substituents independently chosen from hydroxy, amino, oxo, Q-Qaϊkoxy, Ci-C6alkanoyioxy, mono- or di-(Ci-C6alkyl)amino, mono- or di-(Ci-C6aikyl)aminocarbonyl and (4- to 7-membered heterocycioalkylJCo-Cialkyl;
Each R7, R9 and R! 5 is independently hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl, aminosulfonyl, Q-Qalkyl, C2-C6aikenyl, C2-C6alkynyl, Q-Cήalkoxy. Q- C6alkanoyl, C3-C6alkanone, Q-C6alkanoyloxy, C2-C6alky] ether, CrC6alkoxycarbonyl, Q- C6alkylthio, C,-C6alkylsulfonyl, mono- or di-(Ci-C6alkyl)aminoC0-C4alkyl, mono- or di-(Q- C6alkyl)ammocarbonylC0-C4alkyl, mono- or di-(Cj-C6alkyl)aminosulfonyiCo-C4alkyI, Q- Cehaloalkyl, Q-Qhaloalkoxy, C]-C6hydroxyalkyl, CrC6aminoalkyl3 Q-Qcyanoalkyl, or mono- or di-(C,-C6alkyl)amiπoCo-C4alkyl; Or R9 is taken together with Rj to form a fused cycloalkyl or heterocycloalkyi.
1 19. A compound or salt or hydrate thereof according to claim 1 18, wherein RA is 6- to 10- membered aryi or 5- to 10-membered heteroaryl, each of which is substituted with from 0 to 4 substituents independently chosen from RB-
120. A compound or salt or hydrate thereof according to claim 1 19, wherein:

A and E are independently N or CR5;
B, J and D are independently N or CR6;
Each R5 and R6 is independently:
(i) hydrogen, halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosulfonyl; (ii) C,-C6alky], C2-C6alkenyl, CrC6alkynyl, CrC6alkoxy, C,-C6alkanoyl, CrC6alky!thio, Q- C^alkanoyloxy, Cj-Cealkoxycarbonyl, Cj-Cealkylsulfonyl, Cj-Cβhaloalkyl, mono- or di-(Cr C6alkyl)aminoCo-C4alkyl, mono- or di-(Ci-C6alkyl)aminocarbonylC0-C4alkyl, mono- or di- (CrC6alkyl)aminosulfonyICo-C4alkyl, (C3-C7cycIoalkyl)C0-C2alkyl, (4- to 10-membered heterocycle)Co-C2alkyl, phenyiCo-C2alkyl or (3- to 10-membered cycle)C0-C2alkoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano. halogen, amino, oxo, Q^alkyl, C]-C4aikoxy, Q-C^aloalkyl, CrC4haloalkoxy, mono- or di-(Ci-C6alkyl)amino. or 5- or 6-membered heterocycloalkyi.
121. A compound or salt or hydrate thereof according to claim 120, wherein: each Rs is independently:
(i) hydrogen, halogen, nitro, cyano or -COOH; or (ii) Ci-C6aϊky], C2-C6alkenyl, C2-C6alkynyl, CrQalkanoyl, C3-Qalkanone, C2-C6alkyl ether, Cj- C6alkoxycarbony], Ci-C6alkylsulfonyl or  each of which is substituted with from 0 to 3 substituents independently chosen from hydroxy, amino, cyano, Ci-C6alkoxy and mono- or di-(CrC6aIky])amino; and each R^ is independently:
each of which is substituted with from 0 to 3 substituents independently chosen from hydroxy, amino, cyano, Ci-C6alkoxy and mono- or di-(CrC6aIky])amino; and each R^ is independently:
(i) hydrogen, halogen, hydroxy, nitro, cyano or-COOH; or
(ii) CrC6alkyl, C2-C6a!kenyl, C2-C6aikynyl, CrC6alkoxy, CrC6alkanoyl, Ci-Qalkylthio, C,- C6alkanoyloxy, C]-C6alkoxycarbonyl, CrC6aikylsulfonyl, CpQhaioalkyl, (C3- C7cycloaIkyl)Co-C2aikyl, (4- to 10-membered heterocycle)C0-C2aIkyl or phenylCo-C2alkyl; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C]-C4alkyl, Ci-C4aikoxy, Ci-C4haloalkyl, Cp C4haloalkoxy, mono- or di-(C|-C6alkyl)amino and 5- or 6-membered heterocycloalkyl; or two adjacent R^ groups are taken together to form a fused, 5- or 6-membered ring,
122. A compound or salt or hydrate thereof according to claim 121, wherein J or B is substituted carbon.
123. A compound or salt or hydrate thereof according to claim 122, wherein two of B1 J and D are substituted carbon.
124. A compound or salt or hydrate thereof according to claim 1 18, wherein RA is C]- C6alkyl, Ci-C6haloalkyl, (C3-C3cycloalkyl)Co-C4alkyl or (5- to 7-membered heterocycloa!kyl)Co- C4alkyl, each of which is substituted with from 0 to 4 substituents independently chosen from:
(i) halogen, hydroxy, nitro or cyano; and
(ii) C-Qaikyl, C2-C6alkenyl, C2-C6alkynyL Q-Qalkanoyl, C,-C6alkylthio, C]-C6alkanoyloxy, C,-C6a]koxycarbonyl, C,-C6alkylsulfonyl, CrC6haloalkyl, (C3-C7cycIoalkyl)C0-C2aikyl, (4- to 7-membered heterocycle)Co-C2alky] or phenylCo-C2alkyl; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, halogen, amino, oxo, C,- C4alkyl,  CrC4haloalkyl, Ci-C4haloalkoxy, mono- or di-(CrC6alkyl)amino and 5- or 6-membered heterocycloalkyl.
CrC4haloalkyl, Ci-C4haloalkoxy, mono- or di-(CrC6alkyl)amino and 5- or 6-membered heterocycloalkyl.
125. A compound or salt or hydrate thereof according to claim 119, wherein RA is a 5- membered heteroaryl that is substituted with from 0 to 3 substituents independently chosen from:
(i) halogen, hydroxy, nitro, cyano, amino, -COOH, aminocarbonyl or aminosuifσnyl;
(ii) C,-C6alkyl, C2-C6alkenyl, CrC6alkynyl, C,-C6alkoxy, CrC6alkanoyl, CrC6alkylthio, C,- C6alkanoyloxy, CpCfialkoxycarbonyl, C1 -Chalky lsulfonyl, CrC6haloaikyl, mono- or di-(Cr C6alkyl)aminoC0-C4alkyl, mono- or di-(Ci-C6alkyl)aminocarbonylC0-C4alkyl, mono- or di- (C,-C6alkyl)aminosuIfonylCo-C4aϊkyl, (C3-C7cyc!oalkyl)Co-C2alkyl, (4- to 10-membered heterocycle)Co-C2alkyl, phenylCVQalkyl or (3- to ϊ O-membereci cycle)C0-C2alkoxy; each of which is substituted with from 0 to 4 substituents independently chosen from hydroxy, cyano, haiogen, amino, oxo, Ci-C4a!kyl, Ci-C4alkoxy, C,-C4haloa!kyl., Ci-Gjhaloalkoxy, mono- or di-(Ci-Cύaikyl)amino, or 5- or 6-membered heterocycloalkyl.
126. A compound or salt or hydrate thereof according to any one of claims 1 18-125, wherein X is Q=O).
127. A compound or salt or hydrate thereof according to any one of claims 1 18-126, wherein at least one OfR9 and R15 is not H.
128. A compound or salt or hydrate thereof according to claim 127, wherein R9 and R!5 are both other than hydrogen.
129. A compound or salt or hydrate thereof according to claim 128, wherein R9 and RJ S are independently CrC2alkoxy or Cj-C2haloaikyl.
130. A compound or salt or hydrate thereof according to claim 129, wherein R9 and R!5 are each methyl .
131. A compound or salt or hydrate thereof according to claim 129, wherein R9 and R] 5 are independently chosen from CrC2alkyl and each R7 is independently hydrogen, CrC2alkyi, C1- Qalkoxy or CrC2haIoalkyl.
132. A compound or salt or hydrate thereof according to any one of claims 1 18-131, wherein Ri is not H.
133. A compound or salt or hydrate thereof according to claim 132, wherein: Ri is C)-R8; and
R8 is hydrogen, Ci-Cgalkyl, C2-C3alkenyl, C2-C8alkynyl, C2-C8aikyl ether, mono- or di-(CV Csalkyl)aminoC0-C6alkyl, (C.i-C7cycloalkyl)Co-Cβalkyl or (5- to 10-membered heterocyc Ie)CV C6alkyl, each of which is substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosulfonyl, -COOH, CVQalkyl, Ci-C6alkoxy, CrC6haioalky!, CrC6haloalkoxy, mono- or di-(CrC6alkyl)aminoC0-C6alkyl, Cr Cealkylsulfonyl, C]-C6alkylthio, CpCealkylaminosulfonyl, CrC6alkoxycarbonyl or Cp C6alkanoylamino.
134. A compound or salt or hydrate thereof according to claim 133, wherein Rg is hydrogen, CrC6alkyl, C2-C6alkenyl, C2-C6alkynyl, mono- or di-(Ci-Cgalkyl)aminoCrC6alkyl or (5- to 7-membered heterocycle)C0-C(;alkyi, each of which is substituted with from 0 to 3 substituents independently chosen from halogen, amino, hydroxy, cyano, oxo, aminocarbonyl, imino, aminosulfonyl, CrC4alkyI, C]-C4alkoxy and mono- and di-(CrCgalkyI)aminoCo-C6alkyl.
135. A compound or salt or hydrate thereof according to claim 133, wherein Ri is methoxy.
136. A compound or salt or hydrate thereof according to any one of claims 1 18-135, wherein R3 is not H.
137. A compound or salt or hydrate thereof according to claim 136, wherein R3 is methyl.
138. A compound or salt or hydrate thereof, wherein the compound is any one of compounds 1 -1440.
139. A compound or salt or hydrate thereof according to any one of claims 1-138, wherein the compound exhibits no detectable agonist activity an in vitro assay of MCH receptor agonism.
140. A compound or salt or hydrate thereof according to any one of claims 1-139, wherein the compound has an IC50 value of 1 micromolar or less in an in vitro assay of MCH receptor antagonism,
141. A phaπnaceutical composition, comprising a compound or salt or hydrate thereof according to any one of claims 1 to 140, in combination with at least one physiologically acceptable carrier or excipient.
142. A pharmaceutical composition according to claim 141, wherein the composition is formulated as an injectible fluid, an aerosol, a cream, an oral liquid, a tablet, a gel, a pill, a capsule, a syrup, or a transdermal patch.
143. A method for modulating binding of MCH to cellular MCH receptor, the method comprising contacting cells expressing MCH receptor with a compound or salt or hydrate thereof according to any one of claims 1 to 140, in an amount sufficient to detectably modulate MCH binding to MCH receptor in vitro, and thereby modulating MCH binding to MCH receptor in the cells.
144. A method according to claim 143, wherein the cells are present in an animal.
145. A method according to claim 144, wherein animal is a human, the cells are brain cells, and the fluid is cerebrospinal fluid.
146. A method according to claim 143, wherein the modulation is inhibition.
147. A method for modulating binding of MCH to MCH receptor in vitro, the method comprising contacting MCH receptor with a compound or sait or hydrate thereof according to any one of claims 1 to 140, under conditions and in an amount sufficient to detectably modulate MCH binding to the MCH receptor,
148. A method for altering the signal-transducing activity of MCH receptor in a cell, the method comprising contacting a cell expressing MCH receptor with a compound or salt or hydrate thereof according to any one of claims 1 to 140, under conditions and in an amount sufficient to detectably alter the electrophysiology of the cell, and thereby altering the signal-transducing activity of MCH receptor in the cell.
149. A method according to claim 148, wherein the cell is present in an animal.
150. A method according to claim 149, wherein animal is a human, the cell is a brain cell, and the fluid is cerebrospinal fluid.
151. A method according to claim 149, wherein the alteration in the electrophysiology of the ceil is detected as a change in the animal's feeding behavior.
152. A method according to claim 148, wherein the signal-transducing activity of MCH receptor in the cell is inhibited.
153. A method for treating a disease or disorder associated with MCH receptor activation, comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound or salt or hydrate thereof according to any one of claims 1 to 140.
154. A method according to claim 153, wherein the disease or disorder is an eating disorder, sexual disorder, diabetes, metabolic syndrome, heart disease or stroke.
155. A method according to claim 153 or 154, wherein the compound or salt or hydrate thereof is administered oraily.
156. A method according to claim 153 or 154, wherein the compound or salt or hydrate thereof is administered intranasally, intravenously or topically.
157. A method according to any one of claims 153 to 156, wherein the patient is a human.
158. A method according to any one of claims 153 to 156, wherein the patient is a dog or a cat.
159. A method for treating obesity, comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound or salt or hydrate thereof according to any one of claims 1 to 140.
160. A method according to claim 159, wherein the compound or salt or hydrate thereof is administered orally.
161. A method according to claim 159 or 160, wherein the patient is a human.
162. A method according to claim 159 or 160, wherein the patient is a dog or a cat.
163. A compound or salt or hydrate thereof according to any one of claims 1 to 140, wherein the compound is radiolabeled.
] 64. A method for determining the presence or absence of MCH receptor in a sample, comprising: contacting a sample with a compound or salt or hydrate thereof according to any one of claims 1 to 140 under conditions that permit binding of the compound to MCH receptor; and detecting a level of compound or salt or hydrate thereof bound to MCH receptor, and therefrom determining the presence or absence of MCH receptor in the sample.
165. A method according to claim 164, wherein the compound is radiolabeled, and wherein detecting a level of compound comprises: separating unbound compound from bound compound; and determining an amount of bound compound in the sample.
166. A method of claim 164, wherein the sample is a tissue section.
167. A method for treating a patient, comprising diagnosing the patient as having a disease or disorder associated with MCH receptor activation, correlating the diagnosis of a disease or disorder associated with MCH receptor activation with the need for administration of a MCH receptor modulator, and administering to the patient an effective amount of a compound or salt or hydrate thereof according to any one of claims 1 to 140.
168. A packaged pharmaceutical preparation, comprising: (i) a pharmaceutical composition according to claim 141 in a container; and (ii) instructions for using the composition to treat a patient suffering from a disorder associated with MCH receptor activation.
169. A packaged pharmaceutical preparation according to claim 168, wherein the disorder is an eating disorder, a sexual disorder, obesity, metabolic syndrome, diabetes, heart disease or stroke.
170. The use of a compound or salt or hydrate thereof according to any one of claims ϊ to 140 for the manufacture of a medicament for the treatment of a condition responsive to MCH receptor modulation.
171. A use according to claim 170, wherein the condition is obesity, an eating disorder, a sexual disorder, diabetes, metabolic syndrome, heart disease or stroke.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US82083406P | 2006-07-31 | 2006-07-31 | |
| US60/820,834 | 2006-07-31 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008016811A2 true WO2008016811A2 (en) | 2008-02-07 |
| WO2008016811A3 WO2008016811A3 (en) | 2008-10-09 |
Family
ID=38997770
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/074282 WO2008016811A2 (en) | 2006-07-31 | 2007-07-25 | Aminopiperidines and realted compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008016811A2 (en) |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009101917A1 (en) * | 2008-02-13 | 2009-08-20 | Eisai R & D Management Co., Ltd. | Bicycloamine derivative |
| WO2009114173A1 (en) * | 2008-03-14 | 2009-09-17 | Exelixis, Inc. | Azabicyclo [3. 2. i] octyl derivatives as 11 beta-hsdl modulators |
| WO2009119726A1 (en) * | 2008-03-28 | 2009-10-01 | 萬有製薬株式会社 | Diarylmethylamide derivative having antagonistic activity on melanin-concentrating hormone receptor |
| WO2009126806A2 (en) | 2008-04-11 | 2009-10-15 | Janssen Pharmaceutica Nv | Thiazolopyridin-2-yloxy-phenyl and thiazolopyrazin-2-yloxy-phenyl amines as modulators of leukotriene a4 hydrolase |
| WO2009080250A3 (en) * | 2007-12-24 | 2009-11-12 | Syngenta Participations Ag | Insecticidal compounds |
| WO2010050456A1 (en) * | 2008-10-30 | 2010-05-06 | 萬有製薬株式会社 | Piperidine compound having di- or tri-arylmethyl structure |
| WO2010132599A1 (en) | 2009-05-14 | 2010-11-18 | Janssen Pharmaceutica Nv | Compounds with two fused bicyclic heteroaryl moieties as modulators of leukotriene a4 hydrolase |
| US20120214805A1 (en) * | 2009-10-29 | 2012-08-23 | Harris Joel M | Bridged bicyclic piperidine derivatives and methods of use thereof |
| EP2566333A1 (en) * | 2010-05-07 | 2013-03-13 | The Board of Trustees of The Leland Stanford Junior University | Identification of stabilizers of multimeric proteins |
| EP2934514A4 (en) * | 2012-12-21 | 2016-06-08 | Univ Leland Stanford Junior | Transthyretin stabilizers and their use for inhibiting transthyretin amyloidosis and protein-protein interactions |
| US9546156B2 (en) | 2012-11-13 | 2017-01-17 | Array Biopharma Inc. | N-bicyclic aryl,N'-pyrazolyl urea, thiourea, guanidine cyanoguanidine compounds as TrkA kinase inhibitors |
| US9562055B2 (en) | 2011-05-13 | 2017-02-07 | Array Biopharma Inc. | Pyrrolidinyl urea, pyrrolidinyl thiourea and pyrrolidinyl guanidine compounds as TrkA kinase inhibitors |
| JPWO2015046595A1 (en) * | 2013-09-30 | 2017-03-09 | 国立大学法人 東京大学 | Adiponectin receptor activating compound |
| US9609869B2 (en) | 2009-12-01 | 2017-04-04 | Syngenta Crop Protection, Llc | Insecticidal compounds based on isoxazoline derivatives |
| US9708272B2 (en) | 2014-08-29 | 2017-07-18 | Tes Pharma S.R.L. | Inhibitors of α-amino-β-carboxymuconic acid semialdehyde decarboxylase |
| US9790178B2 (en) | 2012-11-13 | 2017-10-17 | Array Biopharma Inc. | Pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9790210B2 (en) | 2012-11-13 | 2017-10-17 | Array Biopharma Inc. | N-(monocyclic aryl),N'-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9809578B2 (en) | 2012-11-13 | 2017-11-07 | Array Biopharma Inc. | Pyrazolyl urea, thiourea, guanidine and cyanoguanidine compounds as trkA kinase inhibitors |
| US9822118B2 (en) | 2012-11-13 | 2017-11-21 | Array Biopharma Inc. | Bicyclic heteroaryl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9828360B2 (en) | 2012-11-13 | 2017-11-28 | Array Biopharma Inc. | Pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9896435B2 (en) | 2012-11-13 | 2018-02-20 | Array Biopharma Inc. | N-pyrrolidinyl,N′-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9969694B2 (en) | 2012-11-13 | 2018-05-15 | Array Biopharma Inc. | N-(arylalkyl)-N′-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9981959B2 (en) | 2012-11-13 | 2018-05-29 | Array Biopharma Inc. | Thiazolyl and oxazolyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US10172959B2 (en) | 2014-08-14 | 2019-01-08 | Mamoun M. Alhamadsheh | Systems for stabilizing and delivering active agents |
| WO2019008507A1 (en) * | 2017-07-03 | 2019-01-10 | Glaxosmithkline Intellectual Property Development Limited | 2-(4-chlorophenoxy)-n-((1 -(2-(4-chlorophenoxy)ethynazetidin-3-yl)methyl)acetamide derivatives and related compounds as atf4 inhibitors for treating cancer and other diseases |
| US10351575B2 (en) | 2012-11-13 | 2019-07-16 | Array Biopharma Inc. | Bicyclic urea, thiourea, guanidine and cyanoguanidine compounds useful for the treatment of pain |
| US10835533B2 (en) | 2014-05-15 | 2020-11-17 | Array Biopharma Inc. | 1 -((3S,4R)-4-(3-fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-yl)-3-(4-methyl-3-(2-methylpyrimidin-5-yl)-1-phenyl-1H-pyrazol-5-yl)urea as a TrkA kinase inhibitor |
| JP2022518439A (en) * | 2019-01-17 | 2022-03-15 | グアンジョウ ミフィア メディカル テクノロジー カンパニー リミテッド | Compounds that target the norepinephrine transporter |
| CN115124473A (en) * | 2022-07-12 | 2022-09-30 | 河北科技大学 | Synthesis method of cimetidine related substance B |
| US20230071039A1 (en) * | 2021-07-26 | 2023-03-09 | Zoetis Services Llc | Serotonin 5-ht2b inhibitory compounds |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5028616A (en) * | 1989-09-05 | 1991-07-02 | G. D. Searle & Co. | N-benzylpiperidine amides |
-
2007
- 2007-07-25 WO PCT/US2007/074282 patent/WO2008016811A2/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5028616A (en) * | 1989-09-05 | 1991-07-02 | G. D. Searle & Co. | N-benzylpiperidine amides |
Cited By (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EA019243B1 (en) * | 2007-12-24 | 2014-02-28 | Зингента Партисипейшнс Аг | Insecticidal compounds |
| US8623896B2 (en) | 2007-12-24 | 2014-01-07 | Syngenta Crop Protection, Llc | Insecticidal compounds |
| AU2008340691C1 (en) * | 2007-12-24 | 2013-08-29 | Syngenta Participations Ag | Insecticidal compounds |
| WO2009080250A3 (en) * | 2007-12-24 | 2009-11-12 | Syngenta Participations Ag | Insecticidal compounds |
| AU2008340691B2 (en) * | 2007-12-24 | 2013-05-02 | Syngenta Participations Ag | Insecticidal compounds |
| WO2009101917A1 (en) * | 2008-02-13 | 2009-08-20 | Eisai R & D Management Co., Ltd. | Bicycloamine derivative |
| US8252810B2 (en) | 2008-02-13 | 2012-08-28 | Eisai R&D Management Co., Ltd. | Bicycloamine derivatives |
| JP2011514357A (en) * | 2008-03-14 | 2011-05-06 | エグゼリクシス, インコーポレイテッド | Azabicyclo [3.2.1] octyl derivatives as 11β-HSD1 modulators |
| WO2009114173A1 (en) * | 2008-03-14 | 2009-09-17 | Exelixis, Inc. | Azabicyclo [3. 2. i] octyl derivatives as 11 beta-hsdl modulators |
| CN101981025A (en) * | 2008-03-28 | 2011-02-23 | 万有制药株式会社 | Diarylmethylamide derivative having antagonistic activity on melanin-concentrating hormone receptor |
| WO2009119726A1 (en) * | 2008-03-28 | 2009-10-01 | 萬有製薬株式会社 | Diarylmethylamide derivative having antagonistic activity on melanin-concentrating hormone receptor |
| WO2009126806A2 (en) | 2008-04-11 | 2009-10-15 | Janssen Pharmaceutica Nv | Thiazolopyridin-2-yloxy-phenyl and thiazolopyrazin-2-yloxy-phenyl amines as modulators of leukotriene a4 hydrolase |
| US7939527B2 (en) | 2008-04-11 | 2011-05-10 | Janssen Pharmaceutica Nv | Thiazolopyridin-2-yloxy-phenyl and thiazolopyrazin-2-yloxy-phenyl amines as modulators of leukotriene A4 hydrolase |
| EP2336125A1 (en) | 2008-04-11 | 2011-06-22 | Janssen Pharmaceutica N.V. | Thiazolopyridin-2-yloxy-phenyl and thiazolopyrazin-2-yloxy-phenyl amines as modulators of leukotriene A4 hydrolase |
| US8357684B2 (en) | 2008-04-11 | 2013-01-22 | Janssen Pharmaceutica Nv | Thyazolopyridin-2-yloxy-phenyl and thiazolopyrazin-2-yloxy-phenyl amines as modulators of leukotriene A4 hydrolase |
| WO2010050456A1 (en) * | 2008-10-30 | 2010-05-06 | 萬有製薬株式会社 | Piperidine compound having di- or tri-arylmethyl structure |
| US8399465B2 (en) | 2009-05-14 | 2013-03-19 | Janssen Pharmaceutica Nv | Compounds with two fused bicyclic heteroaryl moieties as modulators of leukotriene A4 hydrolase |
| WO2010132599A1 (en) | 2009-05-14 | 2010-11-18 | Janssen Pharmaceutica Nv | Compounds with two fused bicyclic heteroaryl moieties as modulators of leukotriene a4 hydrolase |
| US9409918B2 (en) * | 2009-10-29 | 2016-08-09 | Merck Sharp & Dohme Corp. | Bridged bicyclic piperidine derivatives and methods of use thereof |
| US20120214805A1 (en) * | 2009-10-29 | 2012-08-23 | Harris Joel M | Bridged bicyclic piperidine derivatives and methods of use thereof |
| US10206400B2 (en) | 2009-12-01 | 2019-02-19 | Syngenta Participations Ag | Insecticidal compounds based on isoxazoline derivatives |
| US9609869B2 (en) | 2009-12-01 | 2017-04-04 | Syngenta Crop Protection, Llc | Insecticidal compounds based on isoxazoline derivatives |
| US10750745B2 (en) | 2009-12-01 | 2020-08-25 | Syngenta Crop Protection, Llc | Insecticidal compounds based on isoxazoline derivatives |
| US11357231B2 (en) | 2009-12-01 | 2022-06-14 | Syngenta Crop Protection Llc | Insecticidal compounds based on isoxazoline derivatives |
| EP2566333A4 (en) * | 2010-05-07 | 2014-04-02 | Univ Leland Stanford Junior | Identification of stabilizers of multimeric proteins |
| US11337935B2 (en) | 2010-05-07 | 2022-05-24 | The Board Of Trustees Of The Leland Stanford Junior University | Identification of stabilizers of multimeric proteins |
| US9308209B2 (en) | 2010-05-07 | 2016-04-12 | The Board Of Trustees Of The Leland Stanford Junio | Identification of stabilizers of multimeric proteins |
| US8877795B2 (en) | 2010-05-07 | 2014-11-04 | The Board Of Trustees Of The Leland Stanford Junior University | Identification of stabilizers of multimeric proteins |
| US10278929B2 (en) | 2010-05-07 | 2019-05-07 | The Board Of Trustees Of The Leland Stanford Junior University | Identification of stabilizers of multimeric proteins |
| EP2566333A1 (en) * | 2010-05-07 | 2013-03-13 | The Board of Trustees of The Leland Stanford Junior University | Identification of stabilizers of multimeric proteins |
| US10039726B2 (en) | 2010-05-07 | 2018-08-07 | The Board Of Trustees Of The Leland Stanford Junior University | Identification of stabilizers of multimeric proteins |
| US9562055B2 (en) | 2011-05-13 | 2017-02-07 | Array Biopharma Inc. | Pyrrolidinyl urea, pyrrolidinyl thiourea and pyrrolidinyl guanidine compounds as TrkA kinase inhibitors |
| US10323022B2 (en) | 2011-05-13 | 2019-06-18 | Array Biopharma Inc. | Pyrrolidinyl urea, pyrrolidinyl thiourea and pyrrolidinyl guanidine compounds as TrkA kinase inhibitors |
| US9878997B2 (en) | 2011-05-13 | 2018-01-30 | Array Biopharma Inc. | Pyrrolidinyl urea, pyrrolidinyl thiourea and pyrrolidinyl guanidine compounds as TrkA kinase inhibitors |
| US9822118B2 (en) | 2012-11-13 | 2017-11-21 | Array Biopharma Inc. | Bicyclic heteroaryl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US10351575B2 (en) | 2012-11-13 | 2019-07-16 | Array Biopharma Inc. | Bicyclic urea, thiourea, guanidine and cyanoguanidine compounds useful for the treatment of pain |
| US9809578B2 (en) | 2012-11-13 | 2017-11-07 | Array Biopharma Inc. | Pyrazolyl urea, thiourea, guanidine and cyanoguanidine compounds as trkA kinase inhibitors |
| US9896435B2 (en) | 2012-11-13 | 2018-02-20 | Array Biopharma Inc. | N-pyrrolidinyl,N′-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US10889589B2 (en) | 2012-11-13 | 2021-01-12 | Array Biopharma Inc. | Bicyclic urea, thiourea, guanidine and cyanoguanidine compounds useful for the treatment of pain |
| US9969694B2 (en) | 2012-11-13 | 2018-05-15 | Array Biopharma Inc. | N-(arylalkyl)-N′-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9981959B2 (en) | 2012-11-13 | 2018-05-29 | Array Biopharma Inc. | Thiazolyl and oxazolyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9790210B2 (en) | 2012-11-13 | 2017-10-17 | Array Biopharma Inc. | N-(monocyclic aryl),N'-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US10851080B2 (en) | 2012-11-13 | 2020-12-01 | Array Biopharma Inc. | Methods of treatment using pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds |
| US9828360B2 (en) | 2012-11-13 | 2017-11-28 | Array Biopharma Inc. | Pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9790178B2 (en) | 2012-11-13 | 2017-10-17 | Array Biopharma Inc. | Pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9546156B2 (en) | 2012-11-13 | 2017-01-17 | Array Biopharma Inc. | N-bicyclic aryl,N'-pyrazolyl urea, thiourea, guanidine cyanoguanidine compounds as TrkA kinase inhibitors |
| US9913826B2 (en) | 2012-12-21 | 2018-03-13 | The Board Of Trustees Of The Leland Stanford Junior University | Compounds and compositions that bind and stabilize transthyretin and their use for inhibiting transthyretin amyloidosis and protein-protein interactions |
| US10842777B2 (en) | 2012-12-21 | 2020-11-24 | The Board Of Trustees Of The Leland Stanford Junior University | Compounds and compositions that bind and stabilize transthyretin and their use for inhibiting transthyretin amyloidosis and protein-protein interactions |
| US10398681B2 (en) | 2012-12-21 | 2019-09-03 | The Board Of Trustees Of The Leland Stanford Junior University | Compounds and compositions that bind and stabilize transthyretin and their use for inhibiting transthyretin amyloidosis and protein-protein interactions |
| EP2934514A4 (en) * | 2012-12-21 | 2016-06-08 | Univ Leland Stanford Junior | Transthyretin stabilizers and their use for inhibiting transthyretin amyloidosis and protein-protein interactions |
| JPWO2015046595A1 (en) * | 2013-09-30 | 2017-03-09 | 国立大学法人 東京大学 | Adiponectin receptor activating compound |
| EP3053911A4 (en) * | 2013-09-30 | 2017-04-12 | The University of Tokyo | Adiponectin receptor-activating compound |
| US10835533B2 (en) | 2014-05-15 | 2020-11-17 | Array Biopharma Inc. | 1 -((3S,4R)-4-(3-fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-yl)-3-(4-methyl-3-(2-methylpyrimidin-5-yl)-1-phenyl-1H-pyrazol-5-yl)urea as a TrkA kinase inhibitor |
| US10596269B2 (en) | 2014-08-14 | 2020-03-24 | Mamoun M. Alhamadsheh | Delivering enhanced active agents |
| US10772967B2 (en) | 2014-08-14 | 2020-09-15 | Mamoun M. Alhamadsheh | Enhanced anticancer agent |
| US10363318B2 (en) | 2014-08-14 | 2019-07-30 | Mamoun M. Alhamadsheh | Enhanced active agents |
| US10172959B2 (en) | 2014-08-14 | 2019-01-08 | Mamoun M. Alhamadsheh | Systems for stabilizing and delivering active agents |
| US11129902B2 (en) | 2014-08-14 | 2021-09-28 | Mamoun M. Alhamadsheh | Enhanced SN-38 anticancer agent |
| US10513499B2 (en) | 2014-08-29 | 2019-12-24 | Tes Pharma S.R.L. | Inhibitors of alpha-amino-beta-carboxymuconic acid semialdehyde decarboxylase |
| US9708272B2 (en) | 2014-08-29 | 2017-07-18 | Tes Pharma S.R.L. | Inhibitors of α-amino-β-carboxymuconic acid semialdehyde decarboxylase |
| US11254644B2 (en) | 2014-08-29 | 2022-02-22 | Tes Pharma S.R.L. | Inhibitors of alpha-amino-beta-carboxymuconic acid semialdehyde decarboxylase |
| WO2019008507A1 (en) * | 2017-07-03 | 2019-01-10 | Glaxosmithkline Intellectual Property Development Limited | 2-(4-chlorophenoxy)-n-((1 -(2-(4-chlorophenoxy)ethynazetidin-3-yl)methyl)acetamide derivatives and related compounds as atf4 inhibitors for treating cancer and other diseases |
| CN110896634A (en) * | 2017-07-03 | 2020-03-20 | 葛兰素史密斯克莱知识产权发展有限公司 | 2- (4-chlorophenoxy) -N- ((1- (2- (4-chlorophenoxy) ethynylazetidin-3-yl) methyl) acetamide derivatives and related compounds as ATF4 inhibitors for the treatment of cancer and other diseases |
| JP2022518439A (en) * | 2019-01-17 | 2022-03-15 | グアンジョウ ミフィア メディカル テクノロジー カンパニー リミテッド | Compounds that target the norepinephrine transporter |
| JP7386383B2 (en) | 2019-01-17 | 2023-11-27 | ペキン ラド テクノロジー カンパニー リミテッド | Compounds that target the norepinephrine transporter |
| US20230071039A1 (en) * | 2021-07-26 | 2023-03-09 | Zoetis Services Llc | Serotonin 5-ht2b inhibitory compounds |
| CN115124473A (en) * | 2022-07-12 | 2022-09-30 | 河北科技大学 | Synthesis method of cimetidine related substance B |
| CN115124473B (en) * | 2022-07-12 | 2023-11-10 | 河北科技大学 | Method for synthesizing cimetidine related substance B |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008016811A3 (en) | 2008-10-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008016811A2 (en) | Aminopiperidines and realted compounds | |
| CA2440559C (en) | Novel non-imidazole compounds | |
| AU2016372048B2 (en) | Heteroarylhydroxypyrimidinones as agonists of the APJ receptor | |
| CA2482551C (en) | 1-(4-piperidinyl) benzimidazolones as histamine h3 antagonists | |
| JP5201817B2 (en) | Pharmaceutical composition | |
| JP2017522346A (en) | Compounds active against bromodomain | |
| NZ566862A (en) | Diarylamine-containing compounds and compositions, and their use as modulators of C-kit receptors | |
| MX2011000460A (en) | Benzazepine derivatives and their use as hstamine h3 antagonists. | |
| CA2481940A1 (en) | (1-4-piperidinyl) benzimidazole derivatives useful as histamine h3 antagonists | |
| JP2005520791A (en) | N, N'-substituted-1,3-diamino-2-hydroxypropane derivatives | |
| CA2580409A1 (en) | Triazole derivative or salt thereof | |
| KR20010032643A (en) | Anilide derivative, production and use thereof | |
| CA2718038A1 (en) | Azabicyclo [3.2.1] octyl derivatives as 11 beta-hsd1 modulators | |
| AU2014234907B2 (en) | Geminally substituted cyanoethylpyrazolo pyridones as Janus kinase inhibitors | |
| EP2976341A1 (en) | Acyclic cyanoethylpyrazolo pyridones as janus kinase inhibitors | |
| TWI377942B (en) | Inhibitors of c-fms kinase | |
| US20040162278A1 (en) | Triazole compounds useful in therapy | |
| JP2008545756A (en) | 1-methyl-1H-pyrazole-4-carboxamides useful as cancer chemotherapeutic agents | |
| KR20210114972A (en) | Substituted pyrrolidine amide III | |
| EP1749001A1 (en) | 3-piperidinylisochroman-5-ols as dopamine agonists | |
| TW202216141A (en) | Combination of an α2-Adrenoceptor subtype C (alpha-2C) antagonists with a TASK1/3 channel blocker for the treatmentof sleep apnea | |
| AU2017229129B2 (en) | Thiazolidinone compounds and use thereof | |
| US5466691A (en) | Thiophene compound | |
| JP2006526021A (en) | 5H-benzo [D] naphtho [2,1-B] azepine derivatives as selective D1 / D5 receptor antagonists to treat obesity and CNS disorders | |
| JP2006526021A5 (en) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07813321 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| NENP | Non-entry into the national phase in: |
Ref country code: RU |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07813321 Country of ref document: EP Kind code of ref document: A2 |