WO2009016216A1 - Triazole derivatives as scd inhibitors - Google Patents
Triazole derivatives as scd inhibitors Download PDFInfo
- Publication number
- WO2009016216A1 WO2009016216A1 PCT/EP2008/060035 EP2008060035W WO2009016216A1 WO 2009016216 A1 WO2009016216 A1 WO 2009016216A1 EP 2008060035 W EP2008060035 W EP 2008060035W WO 2009016216 A1 WO2009016216 A1 WO 2009016216A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- triazole
- tetrahydro
- carboxamide
- formula
- Prior art date
Links
- 0 *C[n]1nnc(C(Nc2cc(CCNC3)c3cc2)=O)c1* Chemical compound *C[n]1nnc(C(Nc2cc(CCNC3)c3cc2)=O)c1* 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention relates to a novel class of compounds believed to be inhibitors of stearoyl-CoA desaturase (SCD), compositions comprising said compounds, methods of synthesis and uses for such compounds in treating and/or preventing various diseases, including those mediated by SCD enzyme, such as diseases related to elevated lipid levels, cardiovascular disease, diabetes, obesity, metabolic syndrome, skin disorders such as acne, diseases or conditions related to cancer and the treatment of symptoms linked to the production of the amyloid plaque-forming A ⁇ 42 peptide such as Alzheimer's disease and the like.
- SCD stearoyl-CoA desaturase
- Acyl desaturase enzymes catalyze the formation of double bonds in fatty acids derived from either dietary sources or de novo synthesis in the liver. Mammals synthesise at least three fatty acid desaturases of differing chain length that specifically catalyze the addition of double bonds at the delta-9, delta-6, and delta-5 positions.
- Stearoyl-CoA desaturases introduce a double bond in the C9-C10 position of saturated fatty acids.
- the preferred substrates for the enzymes are palmitoyl-CoA (16:0) and stearoyl-CoA (18:0), which are converted to palmitoleoyl-CoA (16:1 ) and oleoyl-CoA (18:1 ), respectively.
- the resulting mono-unsaturated fatty acids may then be employed in the preparation of phospholipids, triglycerides, and cholesteryl esters, in vivo.
- SCD1 A number of mammalian SCD genes have been cloned. For example, two genes have been cloned from rats (SCD1 , SCD2) and four SCD genes have been isolated from mice (SCD1 , 2, 3 and 4). While the basic biochemical roles of SCD has been known in rats and mice since the 1970's (Jeffcoat, R et al., Elsevier Science (1984), VoI 4, pp. 85-1 12; de Antueno, RJ, Lipids (1993), Vol. 28, No. 4, pp. 285-290), it has only recently been directly implicated in human diseases processes.
- SCD1 A single SCD gene, SCD1 , has been characterized in humans. SCD1 is described in Brownlie et al, WO 01/62954. A second human SCD isoform has been identified, and because it bears little sequence homology to known mouse or rat isoforms it has been named human SCD5 or hSCD5 (WO 02/26944).
- inhibition of the activity of SCD in vivo can be used to ameliorate and/or treat one or more diseases such as dyslipidemia, hypoalphalipoproteinemia, hyperbetalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial hypercholesterolemia, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, atherosclerosis, obesity, Type I diabetes, Type Il diabetes, insulin resistance, hyperinsulinaemia, metabolic syndrome; other cardiovascular diseases e.g.
- diseases such as dyslipidemia, hypoalphalipoproteinemia, hyperbetalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial hypercholesterolemia, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, atherosclerosis, obesity, Type I diabetes, Type Il diabetes, insulin resistance, hyperinsulinaemia, metabolic syndrome; other cardiovascular diseases e.g.
- peripheral vascular disease reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, thrombosis; hepatic steatosis, non-alcoholic steatohepatitis (NASH) and other diseases related to accumulation of lipids in the liver.
- NASH non-alcoholic steatohepatitis
- An SCD-mediated disease or condition also includes a disorder of polyunsaturated fatty acid (PUFA) disorder, or a skin disorder, including but not limited to eczema, acne, psoriasis, keloid scar formation or prevention, diseases related to production or secretions from mucous membranes, such as monounsaturated fatty acids, wax esters, and the like (US2006/0205713A1 , WO2007/046868, WO2007/046867).
- PUFA polyunsaturated fatty acid
- SCD has been shown to play a physiological role in cholesterol homeostasis and the de novo biosynthesis of cholesterol esters, triglycerides and wax esters required for normal skin and eyelid function and therefore may be useful in the treatment of acne and other skin conditions (Makoto et al. J of Nutrition (2001 ), 131 (9), 2260-2268, Harrison et al. J of Investigative Dermatology (2007) 127(6), 1309-1317).
- An SCD-mediated disease or condition also includes but is not limited to a disease or condition which is, or is related to cancer, neoplasia, malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like (US2006/0205713A1 , WO2007/046868, WO2007/046867).
- SCD-1 has been identified as playing a role in human tumor cell survival and therefore has potential as an anticancer target (Morgan- Lappe et al. 2007 Cancer Res. 67(9) 4390-4398).
- SCD inhibitors may also be useful for treating, delaying the onset of symptoms, or slowing the progression of symptoms of mild cognitive impairment (MCI), Alzheimer's Disease (AD), cerebral amyloid angiopathy (CAA) or dementia associated with Down Syndrome (DS) and other neurodegenerative diseases characterized by the formation or accumulation of amyloid plaques comprising A ⁇ 42 (US2007/0087363A1 ; Myriad Genetics).
- MCI mild cognitive impairment
- AD Alzheimer's Disease
- CAA cerebral amyloid angiopathy
- DS Down Syndrome
- WO2005/011657 describes certain piperazine derivatives useful for modulating SCD activity.
- the present invention provides a compound of formula (I) for inhibiting SCD activity:
- X represents -CONH- or -NHCO-
- R 1 represents:
- benzothiophene or thiophene wherein the benzothiophene or thiophene is optionally substituted by one, two or three groups independently selected from: -C 1-6 alkyl, -C 1-6 haloalkyl (such as -CF 3 ), or halogen (such as chloro, bromo or fluoro),
- Y represents -CH 2 - or -OCH 2 -
- R 2 represents -H
- R 3 represents -H or -C 1-2 alkyl (such as methyl), or a salt thereof.
- the said compounds have been found to inhibit SCD activity and may therefore be useful in the treatment of SCD-mediated diseases such as diseases or conditions caused by or associated with an abnormal plasma lipid profile including dyslipidemia, hypoalphalipoproteinemia, hyperbetalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial hypercholesterolemia, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, atherosclerosis, obesity, Type I diabetes, Type Il diabetes, insulin resistance, hyperinsulinaemia and metabolic syndrome; other cardiovascular diseases e.g.
- peripheral vascular disease e.g., peripheral vascular disease, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, thrombosis, hepatic steatosis, nonalcoholic steatohepatitis (NASH) and other diseases related to accumulation of lipids in the liver; skin disorders e.g.
- eczema eczema, acne, psoriasis, keloid scar formation or prevention, and diseases related to production or secretions from mucous membranes; cancer, neoplasia, malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like; mild cognitive impairment (MCI), Alzheimer's Disease (AD), cerebral amyloid angiopathy (CAA) or dementia associated with Down Syndrome (DS) and other neurodegenerative diseases characterized by the formation or accumulation of amyloid plaques comprising A ⁇ 42.
- MCI mild cognitive impairment
- AD Alzheimer's Disease
- CAA cerebral amyloid angiopathy
- DS Down Syndrome
- X represents -NHCO-. In another aspect of the invention, X represents -CONH-.
- R 1 represents:
- thiophene or benzothiophene wherein the thiophene or benzothiophene is optionally substituted by one, two or three groups independently selected from:
- -Ci -6 alkyl -Ci -6 haloalkyl (such as -CF 3 ), or halogen (such as chloro, bromo or fluoro).
- R 1 represents:
- thiophene or benzothiophene wherein the thiophene or benzothiophene is optionally substituted by one, two or three groups independently selected from:
- -Ci -6 alkyl -Ci -6 haloalkyl (such as -CF 3 ), or halogen (such as chloro, bromo or fluoro).
- R 1 represents -C 6- ioaryl (such as phenyl or napthyl) optionally substituted by: one, two or three groups independently selected from: (a) -Ci -2 alkyl (such as methyl), -Ci -6 haloalkyl (such as -CF 3 ), halogen (such as chloro, bromo or fluoro), (b) -C 6- ioaryl (such as phenyl) optionally substituted by one, two or three groups selected from: -Ci -3 alkyl or -Ci -6 haloalkyl (such as -CF 3 ).
- R 1 represents phenyl optionally substituted by: one, two or three groups independently selected from:
- phenyl optionally substituted by one, two or three groups selected from: -Ci -3 alkyl or - Ci -6 haloalkyl (such as -CF 3 ).
- R 1 represents phenyl optionally substituted by one, two or three groups independently selected from: -Ci -2 alkyl (such as methyl), -Ci -6 haloalkyl (such as -CF 3 ), halogen (such as chloro, bromo or fluoro) or phenyl.
- R 1 represents phenyl optionally substituted by one or two groups independently selected from: -C 1-2 alkyl (such as methyl), -C 1-6 haloalkyl (such as - CF 3 ), halogen (such as chloro, bromo or fluoro) or phenyl.
- R 1 represents phenyl optionally substituted by one, two or three groups independently selected from: -CH 3 , -CF 3 or halogen (such as chloro, bromo or fluoro).
- R 1 represents phenyl optionally substituted by one or two groups independently selected from: -CH 3 -CF 3 or halogen (such as chloro, bromo or fluoro).
- R 1 represents phenyl optionally substituted by one group independently selected from: -CH 3 -CF 3 or halogen (such as chloro, bromo or fluoro).
- R 1 represents phenyl substituted by two chlorine atoms e.g. at the 3, 4 and 3, 5 positions.
- the invention provides a compound of formula (I), where R 1 represents phenyl substituted by phenyl, such as 1 -phenyl.
- R 1 represents naphthyl optionally substituted by: one, two or three groups independently selected from:
- phenyl optionally substituted by one, two or three groups selected from: -Ci -3 alkyl, -Ci- 6 alkoxy or -C 1-6 haloalkyl (such as -CF 3 ).
- R 1 represents naphthyl.
- R 1 represents thiophene optionally substituted by halogen (such as chloro, bromo or fluoro).
- R 1 represents thiophene substituted by chloro.
- R 1 represents benzothiophene optionally substituted by halogen (such as chloro, bromo or fluoro). In another aspect of the invention, R 1 represents benzothiophene optionally substituted by chloro. In another aspect of the invention, R 1 represents benzothiophene.
- halogen such as chloro, bromo or fluoro
- R 1 represents benzothiophene optionally substituted by chloro. In another aspect of the invention, R 1 represents benzothiophene.
- Y represents -CH 2 - (methylene). In another aspect of the invention, Y represents -OCH 2 -.
- R 3 represents hydrogen, -CH 3 (methyl) or -C 2 H 5 (ethyl). In another aspect of the invention, R 3 represents hydrogen or -CH 3 (methyl). In another aspect of the invention, R 3 represents hydrogen. In another aspect of the invention, R 3 represents - CH 3 (methyl). In another aspect of the invention, R 3 represents -C 2 H 5 (ethyl).
- the invention provides a compound of formula (I) wherein X represents -CONH- and Y represents -CH 2 -.
- Certain compounds of formula (I) may exist in stereoisomeric forms (e.g. they may contain one or more asymmetric carbon atoms). The individual stereoisomers (enantiomers and diastereomers) and mixtures of these are included within the scope of the present invention.
- the invention also extends to conformational isomers of compounds of formula (I) and any geometric (cis and/or trans) isomers of said compounds.
- compounds of formula (I) may exist in tautomeric forms other than that shown in the formula and these are also included within the scope of the present invention.
- racemic compounds of formula (I) may be optionally resolved into their individual enantiomers. Such resolutions may conveniently be accomplished by standard methods known in the art. For example, a racemic compound of formula (I) may be resolved by chiral preparative HPLC.
- alkyl refers to straight or branched hydrocarbon chains containing the specified number of carbon atoms.
- Ci -6 alkyl means a straight or branched alkyl containing at least 1 , and at most 6, carbon atoms.
- alkyl as used herein include, but are not limited to, methyl, ethyl, n-propyl, n-butyl, n-pentyl, n-hexyl, isobutyl, isopropyl, t-butyl and 1 ,1-dimethylpropyl.
- alkyl may include alkylene, for example methylene (-CH 2 -), ethylene (-CH 2 CH 2 -) and propylene (-CH 2 CH 2 CH 2 -).
- halogen refers to a fluorine (fluoro), chlorine (chloro), bromine (bromo) or iodine (iodo) atom.
- haloalkyl refers to an alkyl group having one or more carbon atoms and wherein at least one hydrogen atom is replaced with a halogen atom, for example a trifluoromethyl group and the like.
- C 5-1 oheteroaryl refers to an aromatic cyclic group containing 5 to 10 ring-atoms 1 , 2, 3 or 4 of which are hetero-atoms independently selected from nitrogen, oxygen and sulphur and the remaining ring-atoms are carbon, e.g. benzothiophenyl or thienyl.
- This definition includes both monocyclic and bicyclic ring systems and bicyclic structures at least a portion of which is aromatic.
- aryl means an aromatic carbocyclic moiety.
- the definition includes both monocyclic and bicyclic ring systems and bicyclic structures at least a portion of which is aromatic.
- aromatic, aryl groups include naphthyl, anthryl, phenanthryl, indanyl, indenyl, azulenyl, azulanyl, fluorenyl, phenyl and napthyl, and more specifically phenyl and napthyl.
- C 5-1 oheterocyclyr refers to a cyclic group containing 5 to 10 ring- atoms 1 , 2, 3 or 4 of which are hetero-atoms independently selected from nitrogen, oxygen and sulphur and the remaining ring-atoms are carbon, wherein said cyclic group is saturated, partially or fully unsaturated but, which is not aromatic e.g.
- heterocyclyl and heteroaryl groups include: furyl, thienyl, pyrrolyl, pyrrolinyl, pyrrolidinyl, imidazolyl, dioxolanyl, oxazolyl, thiazolyl, imidazolyl, imidazolinyl, imidazolidinyl, pyrazolyl, pyrazolinyl, pyrazolidinyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyranyl, pyridyl, piperidinyl, homopiperazinyl, dioxanyl, morpholino, dithianyl, thiomorpholino, pyridazinyl, pyrimidinyl, pyrazinyl, piperazinyl, sulfolanyl, tetrazolyl, triazinyl, azepinyl,
- pharmaceutically acceptable means a compound which is suitable for pharmaceutical use.
- Salts and solvates of compounds of formula (I) which are suitable for use in medicine are those wherein the counterion or associated solvent is pharmaceutically acceptable.
- salts and solvates having non-pharmaceutically acceptable counterions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts and solvates.
- Suitable pharmaceutically acceptable salts will be apparent to those skilled in the art and include for example acid addition salts formed with inorganic acids e.g. hydrochloric, hydrobromic, sulfuric, nitric or phosphoric acid; and organic acids e.g. succinic, maleic, malic, mandelic, acetic, fumaric, glutamic, lactic, citric, tartaric, benzoic, benzenesulfonic, p- toluenesulfonic, methanesulfonic, ethanesulfonic or naphthalenesulfonic acid.
- Other non- pharmaceutically acceptable salts e.g. oxalates, may be used, for example in the isolation of compounds of formula (I) and are included within the scope of this invention. Reference is made to Berge et al. J. Pharm. ScL, 1977, 66, 1-19.
- Certain of the compounds of formula (I) may form acid addition salts with one or more equivalents of the acid.
- the present invention includes within its scope all possible stoichiometric and non-stoichiometric forms thereof.
- solvate refers to a complex of variable stoichiometry formed by a solute (in this invention, a compound of formula (I) or a salt thereof) and a solvent.
- solvents for the purpose of the invention may not interfere with the biological activity of the solute.
- suitable solvents include, but are not limited to, water, methanol, ethanol and acetic acid.
- the solvent used is a pharmaceutically acceptable solvent.
- the solvent used is water and the solvate may also be referred to as a hydrate.
- Solvates of compounds of formula (I) which are suitable for use in medicine are those wherein the solvent is pharmaceutically acceptable.
- solvates having non- pharmaceutically acceptable solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts.
- prodrug means a compound which is converted within the body, e.g. by hydrolysis in the blood, into its active form that has medical effects.
- Pharmaceutically acceptable prodrugs are described in T. Higuchi and V. Stella, Prodrugs as Novel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series, and in Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987 and in D. Fleishner, S. Ramon and H. Barba "Improved oral drug delivery: solubility limitations overcome by the use of prodrugs", Advanced Drug Delivery Reviews (1996) 19(2) 1 15-130.
- Prodrugs are any covalently bonded carriers that release a compound of structure (I) in vivo when such prodrug is administered to a patient.
- Prodrugs are generally prepared by modifying functional groups in a way such that the modification is cleaved in vivo yielding the parent compound.
- Prodrugs may include, for example, compounds of this invention wherein hydroxy or amine groups are bonded to any group that, when administered to a patient, cleaves to form the hydroxy or amine groups.
- representative examples of prodrugs include (but are not limited to) phosphonate, carbamate, acetate, formate and benzoate derivatives of hydroxy and amine functional groups of the compounds of formula (I).
- Phosphonates and carbamates may be active in their own right and/or be hydrolysable under in vivo conditions in the human body. Suitable pharmaceutically acceptable in vivo hydrolysable ester groups include those which break down readily in the human body to leave the parent acid or its salt.
- a phosphonate is formed by reaction with phosphorous (phosphonic) acid, by methods well known in the art. For example, phosphonates may be derivatives such as RP(O)(OR) 2 and the like.
- a carbamate is an ester of carbamic acid.
- a compound, or a salt thereof, wherein the compound is selected from the group consisting of:
- the compounds of the invention have been found to inhibit SCD activity and may therefore be useful in regulating lipid levels, e.g. plasma lipid levels.
- Diseases or conditions caused by or associated with an abnormal plasma lipid profile include dyslipidemia, hypoalphalipoproteinemia, hyperbetalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial hypercholesterolemia, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, atherosclerosis, obesity, Type I diabetes, Type Il diabetes, insulin resistance, hyperinsulinaemia and metabolic syndrome.
- Other cardiovascular diseases for which the compounds of the present invention are useful include peripheral vascular disease, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes and thrombosis.
- Other diseases or conditions include hepatic steatosis, non-alcoholic steatohepatitis (NASH) and other diseases related to accumulation of lipids in the liver.
- NASH non-alcoholic steatohepatit
- the compounds of the invention may also be useful in the treatment of skin disorders e.g. eczema, acne, psoriasis, keloid scar formation or prevention, and diseases related to production or secretions from mucous membranes.
- skin disorders e.g. eczema, acne, psoriasis, keloid scar formation or prevention, and diseases related to production or secretions from mucous membranes.
- the compounds of the invention may also be useful in the treatment of cancer, neoplasia, malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like.
- the compounds of the invention may also be useful in the treatment of mild cognitive impairment (MCI), Alzheimer's disease (AD), cerebral amyloid angiopathy (CAA) or dementia associated with Down Syndrome (DS) and other neurodegenerative diseases characterized by the formation or accumulation of amyloid plaques comprising A ⁇ 42.
- MCI mild cognitive impairment
- AD Alzheimer's disease
- CAA cerebral amyloid angiopathy
- DS dementia associated with Down Syndrome
- other neurodegenerative diseases characterized by the formation or accumulation of amyloid plaques comprising A ⁇ 42.
- the terms describing the indications used herein are classified in the Merck Manual of Diagnosis and Therapy, 17 th Edition and/or the International Classification of Diseases 10 th Edition (ICD-10).
- ICD-10 International Classification of Diseases 10 th Edition
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in medical therapy.
- the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating and/or preventing a disease or a condition susceptible to amelioration by an SCD inhibitor.
- the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating and/or preventing acne, cancer, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or nonalcoholic steatohepatitis (NASH).
- a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating and/or preventing acne, cancer, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or nonalcoholic steatohepatitis (NASH).
- NASH nonalcoholic steatohepatitis
- the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating and/or preventing acne, cancer, dyslipidemia, atherosclerosis, insulin resistance, hyperinsulinaemia, Type Il diabetes and/or hepatic steatosis.
- the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating and/or preventing acne.
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in treating and/or preventing a disease or a condition susceptible to amelioration by an SCD inhibitor in a mammal, including human.
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in treating and/or preventing acne, cancer, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or non-alcoholic steatohepatitis (NASH).
- acne cancer, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or non-alcoholic steatohepatitis (NASH).
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in treating and/or preventing acne, cancer, dyslipidemia, atherosclerosis, insulin resistance, hyperinsulinaemia, Type Il diabetes and/or hepatic steatosis.
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in treating and/or preventing acne.
- the invention provides a method for treating and/or preventing a disease or a condition susceptible to amelioration by an SCD inhibitor, which method comprises administering to a subject, for example a mammal, including human, a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the invention provides a method for treating and/or preventing a acne, cancer, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or non-alcoholic steatohepatitis (NASH), which method comprises administering to a subject, for example a mammal, including human, a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- a subject for example a mammal, including human, a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the invention provides a method for treating and/or preventing acne, cancer, dyslipidemia, atherosclerosis, insulin resistance, hyperinsulinaemia, Type Il diabetes and/or hepatic steatosis, which method comprises administering to a subject, for example a mammal, including human, a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the invention provides a method for treating and/or preventing acne, which method comprises administering to a subject, for example a mammal, including human, a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- treatment includes acute treatment or prophylaxis as well as the alleviation of established symptoms.
- the compounds of the invention are intended for use in pharmaceutical compositions it will readily be understood that they are each preferably provided in substantially pure form, for example at least 60% pure, more suitably at least 75% pure and preferably at least 85%, especially at least 98% pure (% are on a weight for weight basis). Impure preparations of the compounds may be used for preparing the more pure forms used in the pharmaceutical compositions; these less pure preparations of the compounds should contain at least 1%, more suitably at least 5% and preferably from 10 to 59% of a compound of the invention.
- final compounds of formula (I) can be converted into other compounds of formula (I) by techniques known to those in the art, for example, carboxylic acid substituents can be converted to esters or amides by routine techniques.
- compounds of formula (I), wherein X represents -CONH- (formula (Ia)) may be prepared according to reaction scheme 1 by reacting compounds of formula (III) and compounds of formula (IV) wherein P 1 represents a suitable nitrogen protecting group such as Boc to form a compound of formula (II).
- the reaction is suitably carried out in the presence of a coupling reagent such as HATU and a base such as DIPEA in a suitable solvent such as DCM or DMF (suitably at room temperature to 80 0 C), and is followed by deprotection of compound of formula (II) under acidic conditions such as hydrochloric acid in a suitable solvent such as ethyl acetate (suitably at room temperature).
- the invention provides a process for the preparation of compounds of the formula (Ia) by reacting compounds of formula (III), wherein R 1 and R 3 are defined above, with compounds of formula (IV), wherein P 1 is defined above, in the presence of a coupling agent, followed by deprotection of compounds of formula (II).
- Compounds of formula (III) may be prepared according to reaction scheme 2 by reacting compounds of formula (V) and compounds of formula (Vl) in the presence of base such as potassium carbonate in a suitable solvent such as DMF or DMSO (suitably at 40 0 C to 8O 0 C), followed by saponification of compounds of formula (VII) in basic conditions such as sodium hydroxide in a suitable solvent, such as ethanol or methanol to reflux.
- base such as potassium carbonate
- a suitable solvent such as DMF or DMSO (suitably at 40 0 C to 8O 0 C)
- saponification of compounds of formula (VII) in basic conditions such as sodium hydroxide in a suitable solvent, such as ethanol or methanol to reflux.
- Compounds of formula (III), wherein R 3 represents H may be prepared according to reaction scheme 3 by reacting compounds of formula (V) and ethyl 2- propynoate (VIII) in a suitable solvent such as ethanol at reflux, followed by saponification of compounds of formula (IX) in basic conditions such as sodium hydroxide in a suitable solvent, such as ethanol or methanol to reflux.
- compounds of formula (I) wherein X represents -NHCO- may be prepared according to reaction scheme 4 by reacting compounds of formula (Xl) and compound of formula (XII) wherein P 1 represents a suitable nitrogen protecting group such as Boc to form a compound of formula (X).
- the reaction is suitably carried out in the presence of a coupling reagent such as HATU and a base such as DIPEA in a suitable solvent such as DCM or DMF (suitably at room temperature to 80 0 C), and is followed by deprotection of compound of formula (X) under acidic conditions such as hydrochloric acid in ethyl acetate (suitably at room temperature).
- the invention provides a process for the preparation of compounds of the formula (Ib) by reacting compounds of formula (Xl), wherein R 1 and R 3 are defined above, with compounds of formula (XII), wherein wherein P 1 is defined above, in the presence of a coupling agent, followed by deprotection of compounds of formula (X).
- Compounds of formula (Xl) may be prepared according to reaction scheme 5 by reacting compounds of formula (XIa) in the presence of bromine and a base such as potassium hydroxide in a suitable solvent such as water (suitably at 4O 0 C to 80 0 C).
- Compounds of formula (XIa) may be prepared according to reaction scheme 6 starting from compounds of formula (III) in the presence of thionyl chloride in chloroform at room temperature followed by reaction with aqueous ammonia in acetonitrile on ice (e.g at -5°C to 5 0 C).
- the compounds of the invention may be prepared singly or as compound libraries comprising at least 2, for example 5 to 1 ,000 compounds, and more preferably 10 to 100 compounds.
- Libraries of compounds of the invention may be prepared by a combinatorial
- a compound library comprising at least 2 compounds of the invention.
- Suitable protecting groups for use according to the present invention are well known to those skilled in the art and may be used in a conventional manner. See, for example, "Protective groups in organic synthesis” by T.W. Greene and P. G. M. Wuts (John Wiley & sons 1991 ) or "Protecting Groups” by PJ. Kocienski (Georg Thieme Verlag 1994).
- suitable amino protecting groups include acyl type protecting groups (e.g.
- aromatic urethane type protecting groups e.g. benzyloxycarbonyl (Cbz) and substituted Cbz
- aliphatic urethane protecting groups e.g. 9-fluorenylmethoxycarbonyl (Fmoc), t-butyloxycarbonyl (Boc), isopropyloxycarbonyl, cyclohexyloxycarbonyl) and alkyl or aralkyl type protecting groups (e.g. benzyl, trityl, chlorotrityl).
- the compounds of formula (I) or pharmaceutically acceptable salt(s) thereof may also be used in combination with other therapeutic agents.
- the invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or pharmaceutically acceptable salt thereof together with one or more further therapeutic agent(s).
- Compounds of the invention may be administered in combination with other therapeutic agents.
- Preferred therapeutic agents are selected from the list: an inhibitor of cholesteryl ester transferase (CETP inhibitors), a HMG-CoA reductase inhibitor, a microsomal triglyceride transfer protein, a peroxisome proliferator-activated receptor activator (PPAR), a bile acid reuptake inhibitor, a cholesterol absorption inhibitor, a cholesterol synthesis inhibitor, a fibrate, niacin, an ion-exchange resin, an antioxidant, an inhibitor of AcylCoA: cholesterol acyltransferase (ACAT inhibitor), a cannabinoid 1 antagonist, a bile acid sequestrant, a corticosteroid, a vitamin D3 derivative, a retinoid, an immunomodulator, an anti androgen, a keratolytic agent, an anti-microbial, a platinum chemotherapeutic, an antimetabolite, hydroxyurea,
- each compound may differ from that when the compound is used alone.
- Appropriate doses will be readily appreciated by those skilled in the art. It will be appreciated that the amount of a compound of the invention required for use in treatment will vary with the nature of the condition being treated and the age and the condition of the patient and will be ultimately at the discretion of the attendant physician or veterinarian.
- compositions comprising a combination as defined above together with at least one pharmaceutically acceptable carrier and/or excipient comprise a further aspect of the invention.
- the individual components of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations by any convenient route.
- either the SCD inhibitor or the second therapeutic agent may be administered first.
- the combination may be administered either in the same or different pharmaceutical composition.
- the two compounds When combined in the same formulation it will be appreciated that the two compounds must be stable and compatible with each other and the other components of the formulation. When formulated separately they may be provided in any convenient formulation, conveniently in such manner as are known for such compounds in the art.
- the invention also includes a pharmaceutical composition comprising one or more compounds of formula (I) or pharmaceutically acceptable salt (s) in combination with one or more excipients.
- the compounds of the invention may be administered in conventional dosage forms prepared by combining a compound of the invention with standard pharmaceutical carriers or diluents according to conventional procedures well known in the art. These procedures may involve mixing, granulating and compressing or dissolving the ingredients as appropriate to the desired preparation.
- compositions of the invention may be formulated for administration by any route, and include those in a form adapted for oral, topical or parenteral administration to mammals including humans.
- compositions may be in the form of tablets, capsules, powders, granules, lozenges, creams or liquid preparations, such as oral or sterile parenteral solutions or suspensions.
- topical formulations of the present invention may be presented as, for instance, dispersions, lotions, creams, gels, pastes, powders, aerosol sprays, syrups or ointments on sponges or cotton applicators, and solutions or suspensions in an aqueous liquid, nonaqueous liquid, oil-in-water emulsion, or water-in-oil liquid emulsion.
- Creams, lotions, or ointments may be prepared as rinse-off or leave-on products, as well as two stage treatment products for use with other skin cleansing or managing compositions.
- the compositions can be administered as a rinse-off product in a higher concentration form, such as a gel, and then a leave-on product in a lower concentration to avoid irritation of the skin.
- Ointments are hydrocarbon-based semisolid formulations containing dissolved or suspended drugs.
- Creams and lotions are semi-solid emulsion systems and the term is applied both to water/oil or oil/water. Gel formulations are semi-solid systems in which a liquid phase is trapped in a polymeric matrix.
- the ointments may contain one or more hydrophobic carriers selected from, for example, white soft paraffin or other mineral waxes, liquid paraffin, non- mineral waxes, long chain alcohols, long chain acids and silicones.
- the ointment may contain in addition to the hydrophobic carriers some hydrophillic carriers selected from, for example, propylene glycol and polyethylene glycol in combination with an appropriate surfactant/co-surfactant system.
- the carrier compositions of the creams or lotions are typically based on water, white soft paraffin and an appropriate surfactant/co-surfactant system, in combination with other carriers/components selected from, for example, propylene glycol, butylene glycol glycerinemonostearate, PEG-glycerinemonostearate, esters such as Ci 2 -i5 alkyl benzoate, liquid paraffin, non-mineral waxes, long chain alcohols, long chain acids silicones, non-silicone polymers.
- an appropriate surfactant/co-surfactant system selected from, for example, propylene glycol, butylene glycol glycerinemonostearate, PEG-glycerinemonostearate, esters such as Ci 2 -i5 alkyl benzoate, liquid paraffin, non-mineral waxes, long chain alcohols, long chain acids silicones, non-silicone polymers.
- the gels may by way of example be formulated using isopropyl alcohol or ethyl alcohol, propylene glycol and water with a gelling agent such as hydroxyethyl cellulose, suitably in combination with minor components, for example one or more of butylene glycol and a wetting agent such as a poloxamer.
- a gelling agent such as hydroxyethyl cellulose
- An ointment, cream, lotion, gel, and the like can further comprise a moisturizing agent.
- the moisturizing agent can be a hydrophobic moisturizing agent such as ceramide, borage oil, tocopherol, tocopherol linoleate, dimethicone or a mixture thereof or a hydrophilic moisturizing agent such as glycerine, hyaluronic acid, sodium peroxylinecarbolic acid, wheat protein, hair keratin amino acids, or a mixture thereof.
- compositions according to the invention may also comprise conventional additives and adjuvants for dermatological applications, such as preservatives, acids or bases used as pH buffer excipients and antioxidants.
- the present invention encompasses administration via a transdermal patch or other forms of transdermal administration.
- Suitable formulations for transdermal administration are known in the art, and may be employed in the methods of the present invention.
- suitable transdermal patch formulations for the administration of a pharmaceutical compound are described in, for example, U.S. Pat. No. 4, 460,372 to Campbell et al., U.S. Pat. No. 4,573,996 to Kwiatek et al., U. S. Pat. No. 4,624,665 to Nuwayser, U.S. Pat. No. 4,722,941 to Eckert et al., and U.S. Pat. No. 5, 223,261 to Nelson et al.
- a suitable transdermal patch for use in the methods of the present invention encompasses a suitable transdermal patch includes a backing layer which is non-permeable, a permeable surface layer, an adhesive layer substantially continuously coating the permeable surface layer, and a reservoir located or sandwiched between the backing layer and the permeable surface layer such that the backing layer extends around the sides of the reservoir and is joined to the permeable surface layer at the edges of the permeable surface layer.
- the reservoir contains a compound of formula (I) or pharmaceutically acceptable salt thereof, alone or in combination, and is in fluid contact with the permeable surface layer.
- the transdermal patch is adhered to the skin by the adhesive layer on the permeable surface layer, such that the permeable surface layer is in substantially continuous contact with the skin when the transdermal patch is adhered to the skin. While the transdermal patch is adhered to the skin of the subject, the compound of formula (I) or pharmaceutically acceptable salt thereof contained in the reservoir of the transdermal patch is transferred via the permeable surface layer, from the reservoir, through the adhesive layer, and to the skin of the patient.
- the transdermal patch may optionally also include one or more penetration- enhancing agents in the reservoir that enhance the penetration of the compound of formula (I) or pharmaceutically acceptable salt thereof through the skin.
- suitable materials which may comprise the backing layer are well known in the art of transdermal patch delivery, and any conventional backing layer material may be employed in the transdermal patch of the instant invention.
- Suitable penetration-enhancing agents are well known in the art as well.
- conventional penetration-enhancing agents include alkanols such as ethanol, hexanol, cyclohexanol, and the like, hydrocarbons such as hexane, cyclohexaue, isopropylbenzene; aldebydes and ketones such as cyclohexanone, acetamide, N,N-di(lower alkyl)acetamides such as N,N-diethylacetamide, N,N-dimethyl acetamide, N-(2-hydroxyethyl) acetamide, esters such as N,N-di-lower alkyl sulfoxides; essential oils such as propylene glycol, glycerine, glycerol monolaurate, isopropyl myristate, and ethyl oleate, salicylates, and mixtures of any of the above.
- Tablets and capsules for oral administration may be in unit dose presentation form, and may contain conventional excipients such as binding agents, for example syrup, acacia, gelatin, sorbitol, tragacanth, or polyvinylpyrrolidone; fillers, for example lactose, sugar, maize-starch, calcium phosphate, sorbitol or glycine; tabletting lubricants, for example magnesium stearate, talc, polyethylene glycol or silica; disintegrants, for example potato starch; or acceptable wetting agents such as sodium lauryl sulphate.
- the tablets may be coated according to methods well known in normal pharmaceutical practice.
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives, such as suspending agents, for example sorbitol, methyl cellulose, glucose syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminium stearate gel or hydrogenated edible fats, emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which may include edible oils), for example almond oil, oily esters such as glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and, if desired, conventional flavouring or colouring agents.
- suspending agents for example sorbitol, methyl cellulose, glucose syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminium stearate gel or hydrogenated edible fats, emulsifying agents, for example lecithin, sorbitan monooleate, or
- Suppositories will contain conventional suppository bases, e.g. cocoa-butter or other glyceride.
- fluid unit dosage forms are prepared utilising the compound and a sterile vehicle, water being preferred.
- the compound depending on the vehicle and concentration used, can be either suspended or dissolved in the vehicle.
- the compound can be dissolved in water for injection and filter sterilised before filling into a suitable vial or ampoule and sealing.
- agents such as a local anaesthetic, preservative and buffering agents can be dissolved in the vehicle.
- the composition can be frozen after filling into the vial and the water removed under vacuum.
- the dry lyophilised powder is then sealed in the vial and an accompanying vial of water for injection may be supplied to reconstitute the liquid prior to use.
- Parenteral suspensions are prepared in substantially the same manner except that the compound is suspended in the vehicle instead of being dissolved and sterilisation cannot be accomplished by filtration.
- the compound can be sterilised by exposure to ethylene oxide before suspending in the sterile vehicle.
- a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
- compositions may contain from 0.1 % by weight, preferably from 10-60% by weight, of the active ingredient, depending on the method of administration. Where the compositions comprise dosage units, each unit will preferably contain from 50-500 mg of the active ingredient.
- the dosage as employed for adult human treatment will preferably range from
- Such a dosage corresponds to 1.5 to 50 mg/kg per day.
- the dosage is from 5 to 20 mg/kg per day.
- the optimal quantity and spacing of individual dosages of a compound of the invention will be determined by the nature and extent of the condition being treated, the form, route and site of administration, and the particular mammal being treated, and that such optimums can be determined by conventional techniques. It will also be appreciated by one of skill in the art that the optimal course of treatment, i.e., the number of doses of a compound of the invention given per day for a defined number of days, can be ascertained by those skilled in the art using conventional course of treatment determination tests.
- Analytical HPLC was conducted on a X-terra MS C18 column (2.5 ⁇ m 3 x 30 mm id) eluting with 0.01 M ammonium acetate in water (solvent A) and 100% acetonitrile using the following elution gradient: 0 to 4 minutes, 5 to 100%B; 4 to 5 minutes, 100%B at a flow-rate of 1.1 mL/min with a temperature of 40 0 C.
- MS mass spectra
- Analytical HPLC was conducted on an Uptisphere-hsc column (3 ⁇ m 30 x 3 mm id) eluting with 0,01 M ammonium acetate in water (solvent A) and 100% acetonitrile (solvent B) using the following elution gradient: 0 to 0.5 minutes, 5%B; 0.5 to 3.5 minutes, 5 to 100%B; 3.5 to 4 minutes, 100%B; 4 to 4.5 minutes, 100 to 5%B; 4.5 to 5.5 minutes, 5%B at a flow-rate of 1.3 mL/min with a temperature of 40 0 C.
- MS mass spectra
- Analytical GC was conducted on a DB-1 ms column (Agilent Technologies), 0.1 ⁇ m 10m x 0.1 mm id) eluting with an Helium flow of 0.5ml/min and pressure at 3.4 bar and with a gradient temperature: 0 to 0.35 min, 100 0 C; 0.35min to 6min, 100 0 C to 250°C (ramp of 80°C/min).
- MS mass spectra
- Example 1 1-(2-Biphenylylmethyl)-5-methyl- ⁇ /-(1 ,2,3,4-tetrahvdro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide hydrochloride
- Example 22 1 -r(3.4-Dichlorophenyl)methyll- ⁇ /-(1.2.3.4-tetrahvdro-6-iso ⁇ uinolinyl)-1 H-1.2.3- triazole-4-carboxamide hydrochloride
- Example 22 was similarly prepared by a method analogous to that described for Example 22:
- the compounds of the present invention may be analysed in vitro for SCD activity using an assay based on the production of [ 3 H]H 2 O, which is released during the enzyme-catalyzed generation of the monounsaturated fatty acyl CoA product.
- the assay is performed in a 96- well filtration plates.
- the titrated substrate used in the assay is the [9,10- 3 H] stearoyl Coenzyme A.
- SCD-containing rat microsomes (2 ⁇ g protein) and substrate (1 ⁇ M) After incubation for 6 minutes of SCD-containing rat microsomes (2 ⁇ g protein) and substrate (1 ⁇ M), the labelled fatty acid acyl-CoA species and microsomes are absorbed with charcoal and separated from [ 3 H]H 2 O by centrifugation.
- [ 3 H]H 2 O is used as a measure of SCD activity.
- Compounds at concentrations starting at 10 ⁇ M to 0.1 nM or vehicle (DMSO) are preincubated for 5 minutes with the microsomes before addition of the substrate.
- the concentration-responses are fitted with sigmoidal curves to obtain IC 50 values.
Abstract
The present invention relates to substituted triazole compounds of the formula (I): and salts thereof, to pharmaceutical compositions containing them and their use in medicine. In particular, the invention relates to compounds for inhibiting SCD activity.
Description
TRIAZOLE DERIVATIVES AS SCD INHIBITORS
FIELD OF THE INVENTION
The present invention relates to a novel class of compounds believed to be inhibitors of stearoyl-CoA desaturase (SCD), compositions comprising said compounds, methods of synthesis and uses for such compounds in treating and/or preventing various diseases, including those mediated by SCD enzyme, such as diseases related to elevated lipid levels, cardiovascular disease, diabetes, obesity, metabolic syndrome, skin disorders such as acne, diseases or conditions related to cancer and the treatment of symptoms linked to the production of the amyloid plaque-forming Aβ42 peptide such as Alzheimer's disease and the like.
BACKGROUND OF THE INVENTION
Acyl desaturase enzymes catalyze the formation of double bonds in fatty acids derived from either dietary sources or de novo synthesis in the liver. Mammals synthesise at least three fatty acid desaturases of differing chain length that specifically catalyze the addition of double bonds at the delta-9, delta-6, and delta-5 positions. Stearoyl-CoA desaturases (SCDs) introduce a double bond in the C9-C10 position of saturated fatty acids. The preferred substrates for the enzymes are palmitoyl-CoA (16:0) and stearoyl-CoA (18:0), which are converted to palmitoleoyl-CoA (16:1 ) and oleoyl-CoA (18:1 ), respectively. The resulting mono-unsaturated fatty acids may then be employed in the preparation of phospholipids, triglycerides, and cholesteryl esters, in vivo.
A number of mammalian SCD genes have been cloned. For example, two genes have been cloned from rats (SCD1 , SCD2) and four SCD genes have been isolated from mice (SCD1 , 2, 3 and 4). While the basic biochemical roles of SCD has been known in rats and mice since the 1970's (Jeffcoat, R et al., Elsevier Science (1984), VoI 4, pp. 85-1 12; de Antueno, RJ, Lipids (1993), Vol. 28, No. 4, pp. 285-290), it has only recently been directly implicated in human diseases processes.
A single SCD gene, SCD1 , has been characterized in humans. SCD1 is described in Brownlie et al, WO 01/62954. A second human SCD isoform has been identified, and because it bears little sequence homology to known mouse or rat isoforms it has been named human SCD5 or hSCD5 (WO 02/26944).
Whilst not wishing to be bound by theory, it is thought that inhibition of the activity of SCD in vivo can be used to ameliorate and/or treat one or more diseases such as dyslipidemia, hypoalphalipoproteinemia, hyperbetalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial hypercholesterolemia, angina, ischemia, cardiac ischemia,
stroke, myocardial infarction, atherosclerosis, obesity, Type I diabetes, Type Il diabetes, insulin resistance, hyperinsulinaemia, metabolic syndrome; other cardiovascular diseases e.g. peripheral vascular disease, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, thrombosis; hepatic steatosis, non-alcoholic steatohepatitis (NASH) and other diseases related to accumulation of lipids in the liver.
An SCD-mediated disease or condition also includes a disorder of polyunsaturated fatty acid (PUFA) disorder, or a skin disorder, including but not limited to eczema, acne, psoriasis, keloid scar formation or prevention, diseases related to production or secretions from mucous membranes, such as monounsaturated fatty acids, wax esters, and the like (US2006/0205713A1 , WO2007/046868, WO2007/046867). SCD has been shown to play a physiological role in cholesterol homeostasis and the de novo biosynthesis of cholesterol esters, triglycerides and wax esters required for normal skin and eyelid function and therefore may be useful in the treatment of acne and other skin conditions (Makoto et al. J of Nutrition (2001 ), 131 (9), 2260-2268, Harrison et al. J of Investigative Dermatology (2007) 127(6), 1309-1317).
An SCD-mediated disease or condition also includes but is not limited to a disease or condition which is, or is related to cancer, neoplasia, malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like (US2006/0205713A1 , WO2007/046868, WO2007/046867). Recently, SCD-1 has been identified as playing a role in human tumor cell survival and therefore has potential as an anticancer target (Morgan- Lappe et al. 2007 Cancer Res. 67(9) 4390-4398).
It has been shown that overexpression of Steroyl-CoA desaturase (SCD) in human cells in culture leads to a specific increase in the production of the amyloid plaque-forming peptide Aβ42, and conversely, that reductions in SCD activity in human cells in culture leads to a specific decrease in the production of Aβ42. Therefore, SCD inhibitors may also be useful for treating, delaying the onset of symptoms, or slowing the progression of symptoms of mild cognitive impairment (MCI), Alzheimer's Disease (AD), cerebral amyloid angiopathy (CAA) or dementia associated with Down Syndrome (DS) and other neurodegenerative diseases characterized by the formation or accumulation of amyloid plaques comprising Aβ42 (US2007/0087363A1 ; Myriad Genetics).
WO2005/011657 describes certain piperazine derivatives useful for modulating SCD activity.
The present invention provides a compound of formula (I) for inhibiting SCD activity:

(I) wherein:
X represents -CONH- or -NHCO-,
R1 represents:
(i) a substituent selected from: -H or -C1-6alkyl, (ii) -C6-ioaryl (such as phenyl or naphthyl) optionally substituted by one, two or three groups independently selected from:
(a) -C1-2alkyl (such as methyl), -C1-6haloalkyl (such as -CF3) or halogen (such as chloro, bromo or fluoro),
(b) -C6-ioaryl (such as phenyl), -C5-1oheteroaryl or -Cs-ioheterocyclyl, wherein the -C6- 10aryl, -C5-1oheteroaryl or -Cs-ioheterocyclyl ring is optionally substituted by one, two or three groups independently selected from: -Ci-3alkyl, -Ci-6alkoxy, or -Ci- 6haloalkyl (such as -CF3),
(iii) benzothiophene or thiophene wherein the benzothiophene or thiophene is optionally substituted by one, two or three groups independently selected from: -C1-6alkyl, -C1-6haloalkyl (such as -CF3), or halogen (such as chloro, bromo or fluoro),
Y represents -CH2- or -OCH2-, R2 represents -H,
R3 represents -H or -C1-2alkyl (such as methyl), or a salt thereof.
The said compounds have been found to inhibit SCD activity and may therefore be useful in the treatment of SCD-mediated diseases such as diseases or conditions caused by or associated with an abnormal plasma lipid profile including dyslipidemia, hypoalphalipoproteinemia, hyperbetalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial hypercholesterolemia, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, atherosclerosis, obesity, Type I diabetes, Type Il diabetes, insulin resistance, hyperinsulinaemia and metabolic syndrome; other cardiovascular diseases e.g. peripheral vascular disease, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, thrombosis, hepatic steatosis, nonalcoholic steatohepatitis (NASH) and other diseases related to accumulation of lipids in the liver; skin disorders e.g. eczema, acne, psoriasis, keloid scar formation or prevention, and diseases related to production or secretions from mucous membranes; cancer, neoplasia,
malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like; mild cognitive impairment (MCI), Alzheimer's Disease (AD), cerebral amyloid angiopathy (CAA) or dementia associated with Down Syndrome (DS) and other neurodegenerative diseases characterized by the formation or accumulation of amyloid plaques comprising Aβ42.
In one aspect of the invention, X represents -NHCO-. In another aspect of the invention, X represents -CONH-.
In one aspect of the invention
R1 represents:
(i) a substituent selected from: -H or -Ci-6alkyl,
(ii) -C6-ioaryl (such as phenyl or naphthyl) optionally substituted by -C1-6haloalkyl (such as -
CF3), (iii) -C6-ioaryl (such as phenyl or naphthyl) optionally substituted by one, two or three groups independently selected from:
(a) -Ci-2alkyl (such as methyl) or halogen (such as chloro, bromo or fluoro),
(b) -C6-ioaryl (such as phenyl), -C5-ioheteroaryl or -Cs-ioheterocyclyl, wherein the -C6- 10aryl, -C5-1oheteroaryl or -Cs-ioheterocyclyl ring is optionally substituted by one, two or three groups independently selected from: -Ci-3alkyl, -Ci-6alkoxy or -Ci- 6haloalkyl (such as -CF3),
(iv) thiophene or benzothiophene wherein the thiophene or benzothiophene is optionally substituted by one, two or three groups independently selected from:
-Ci-6alkyl, -Ci-6haloalkyl (such as -CF3), or halogen (such as chloro, bromo or fluoro).
In another aspect of the invention
R1 represents:
(i) -C6-ioaryl (such as phenyl or naphthyl) optionally substituted by -C1-6haloalkyl (such as -
CF3), (ii) -C6-ioaryl (such as phenyl or naphthyl) optionally substituted by one, two or three groups independently selected from:
(a) -Ci-2alkyl (such as methyl) or halogen (such as chloro, bromo or fluoro),
(b) -C6-ioaryl (such as phenyl), wherein the -C6-ioaryl is optionally substituted by one, two or three groups independently selected from: -C1-3alkyl, -C1-6alkoxy or -C1- 6haloalkyl (such as -CF3),
(iv) thiophene or benzothiophene wherein the thiophene or benzothiophene is optionally substituted by one, two or three groups independently selected from:
-Ci-6alkyl, -Ci-6haloalkyl (such as -CF3), or halogen (such as chloro, bromo or fluoro).
In another aspect of the invention, R1 represents -C6-ioaryl (such as phenyl or napthyl) optionally substituted by: one, two or three groups independently selected from: (a) -Ci-2alkyl (such as methyl), -Ci-6haloalkyl (such as -CF3), halogen (such as chloro, bromo or fluoro),
(b) -C6-ioaryl (such as phenyl) optionally substituted by one, two or three groups selected from: -Ci-3alkyl or -Ci-6haloalkyl (such as -CF3).
In another aspect of the invention, R1 represents phenyl optionally substituted by: one, two or three groups independently selected from:
(a) -Ci-2alkyl (such as methyl), -Ci-6haloalkyl (such as -CF3), halogen (such as chloro, bromo or fluoro),
(b) phenyl optionally substituted by one, two or three groups selected from: -Ci-3alkyl or - Ci-6haloalkyl (such as -CF3).
In another aspect of the invention, R1 represents phenyl optionally substituted by one, two or three groups independently selected from: -Ci-2alkyl (such as methyl), -Ci-6haloalkyl (such as -CF3), halogen (such as chloro, bromo or fluoro) or phenyl.
In another aspect of the invention, R1 represents phenyl optionally substituted by one or two groups independently selected from: -C1-2alkyl (such as methyl), -C1-6haloalkyl (such as - CF3), halogen (such as chloro, bromo or fluoro) or phenyl.
In another aspect of the invention, R1 represents phenyl optionally substituted by one, two or three groups independently selected from: -CH3, -CF3 or halogen (such as chloro, bromo or fluoro).
In another aspect of the invention, R1 represents phenyl optionally substituted by one or two groups independently selected from: -CH3 -CF3 or halogen (such as chloro, bromo or fluoro).
In another aspect of the invention, R1 represents phenyl optionally substituted by one group independently selected from: -CH3 -CF3 or halogen (such as chloro, bromo or fluoro).
In another aspect of the invention, R1 represents phenyl substituted by two chlorine atoms e.g. at the 3, 4 and 3, 5 positions.
In another aspect the invention provides a compound of formula (I), where R1 represents phenyl substituted by phenyl, such as 1 -phenyl.
In another aspect of the invention, R1 represents naphthyl optionally substituted by: one, two or three groups independently selected from:
(a) -C1-2alkyl (such as methyl), -C1-6haloalkyl (such as -CF3), halogen (such as chloro, fluoro or bromo)
(b) phenyl optionally substituted by one, two or three groups selected from: -Ci-3alkyl, -Ci- 6alkoxy or -C1-6haloalkyl (such as -CF3).
In another aspect of the invention, R1 represents naphthyl.
In another aspect of the invention, R1 represents thiophene optionally substituted by halogen (such as chloro, bromo or fluoro). In another aspect of the invention, R1 represents thiophene substituted by chloro.
In another aspect of the invention, R1 represents benzothiophene optionally substituted by halogen (such as chloro, bromo or fluoro). In another aspect of the invention, R1 represents benzothiophene optionally substituted by chloro. In another aspect of the invention, R1 represents benzothiophene.
In another aspect of the invention, Y represents -CH2- (methylene). In another aspect of the invention, Y represents -OCH2-.
In one aspect of the invention, R3 represents hydrogen, -CH3 (methyl) or -C2H5 (ethyl). In another aspect of the invention, R3 represents hydrogen or -CH3 (methyl). In another aspect of the invention, R3 represents hydrogen. In another aspect of the invention, R3 represents - CH3 (methyl). In another aspect of the invention, R3 represents -C2H5 (ethyl).
Each of the aspects of the invention are independent unless stated otherwise. Nevertheless the skilled person will understand that all the permutations of the aspects of described are within the scope of the invention. Thus it is to be understood that the present invention covers all combinations of suitable, convenient and exemplified groups described herein. For example, in one aspect the invention provides a compound of formula (I) wherein X represents -CONH- and Y represents -CH2-.
Certain compounds of formula (I) may exist in stereoisomeric forms (e.g. they may contain one or more asymmetric carbon atoms). The individual stereoisomers (enantiomers and diastereomers) and mixtures of these are included within the scope of the present invention. The invention also extends to conformational isomers of compounds of formula (I) and any geometric (cis and/or trans) isomers of said compounds. Likewise, it is understood that compounds of formula (I) may exist in tautomeric forms other than that shown in the formula and these are also included within the scope of the present invention.
It will be appreciated that racemic compounds of formula (I) may be optionally resolved into their individual enantiomers. Such resolutions may conveniently be accomplished by standard methods known in the art. For example, a racemic compound of formula (I) may be resolved by chiral preparative HPLC.
It will also be appreciated that compounds of the invention which exist as polymorphs, and mixtures thereof, are within the scope of the present invention.
As used herein, the term "alkyl" refers to straight or branched hydrocarbon chains containing the specified number of carbon atoms. For example, Ci-6alkyl means a straight or branched alkyl containing at least 1 , and at most 6, carbon atoms. Examples of "alkyl" as used herein
include, but are not limited to, methyl, ethyl, n-propyl, n-butyl, n-pentyl, n-hexyl, isobutyl, isopropyl, t-butyl and 1 ,1-dimethylpropyl. However, when a moiety is defined such that alkyl bears a substituent it will be clear to the skilled person from the context that alkyl may include alkylene, for example methylene (-CH2-), ethylene (-CH2CH2-) and propylene (-CH2CH2CH2-).
As used herein, the term "halogen" or "halo" refers to a fluorine (fluoro), chlorine (chloro), bromine (bromo) or iodine (iodo) atom.
As used herein, the term "haloalkyl" refers to an alkyl group having one or more carbon atoms and wherein at least one hydrogen atom is replaced with a halogen atom, for example a trifluoromethyl group and the like.
As used herein, the term "C5-1oheteroaryl" refers to an aromatic cyclic group containing 5 to 10 ring-atoms 1 , 2, 3 or 4 of which are hetero-atoms independently selected from nitrogen, oxygen and sulphur and the remaining ring-atoms are carbon, e.g. benzothiophenyl or thienyl. This definition includes both monocyclic and bicyclic ring systems and bicyclic structures at least a portion of which is aromatic.
As used herein, the term "aryl" means an aromatic carbocyclic moiety. The definition includes both monocyclic and bicyclic ring systems and bicyclic structures at least a portion of which is aromatic. Examples of aromatic, aryl groups include naphthyl, anthryl, phenanthryl, indanyl, indenyl, azulenyl, azulanyl, fluorenyl, phenyl and napthyl, and more specifically phenyl and napthyl.
As used herein, the term "C5-1oheterocyclyr refers to a cyclic group containing 5 to 10 ring- atoms 1 , 2, 3 or 4 of which are hetero-atoms independently selected from nitrogen, oxygen and sulphur and the remaining ring-atoms are carbon, wherein said cyclic group is saturated, partially or fully unsaturated but, which is not aromatic e.g. tetrahydrofuran, dihydrofuran, 1 ,4- dioxane, morpholine, 1 ,4-dithiane, piperazine, piperidine, 1 ,3-dioxolane, imidazolidine, imidazoline, pyrroline, pyrrolidine, tetrahydropyran, dihydropyran, oxathiolane, 1 ,3-dioxane, 1 ,3-dithiane, oxathiane or thiomorpholine. This definition includes bicyclic structures provided the moiety is non-aromatic.
Examples of heterocyclyl and heteroaryl groups include: furyl, thienyl, pyrrolyl, pyrrolinyl, pyrrolidinyl, imidazolyl, dioxolanyl, oxazolyl, thiazolyl, imidazolyl, imidazolinyl, imidazolidinyl, pyrazolyl, pyrazolinyl, pyrazolidinyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyranyl, pyridyl, piperidinyl, homopiperazinyl, dioxanyl, morpholino, dithianyl, thiomorpholino, pyridazinyl, pyrimidinyl, pyrazinyl, piperazinyl, sulfolanyl, tetrazolyl, triazinyl, azepinyl, oxazepinyl, thiazepinyl, diazepinyl and thiazolinyl, benzimidazolyl, benzoxazolyl, imidazopyridinyl, benzoxazinyl, benzothiazinyl, benzothiophenyl oxazolopyridinyl, benzofuranyl, quinolinyl, quinazolinyl, quinoxalinyl, dihydroquinazolinyl, benzothiazolyl, phthalimido, benzofuranyl, benzodiazepinyl, indolyl and isoindolyl.
As used herein, the term "substituted" refers to substitution with the named substituent or substituents, multiple degrees of substitution being allowed unless otherwise stated.
For the avoidance of doubt, the term "independently" means that where more than one substituent is selected from a number of possible substituents, those substituents may be the same or different.
In one aspect of the invention there is provided a compound of formula (I) or a pharmaceutically acceptable salt thereof.
As used herein, the term "pharmaceutically acceptable" means a compound which is suitable for pharmaceutical use.
Salts and solvates of compounds of formula (I) which are suitable for use in medicine are those wherein the counterion or associated solvent is pharmaceutically acceptable. However, salts and solvates having non-pharmaceutically acceptable counterions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts and solvates.
Suitable pharmaceutically acceptable salts will be apparent to those skilled in the art and include for example acid addition salts formed with inorganic acids e.g. hydrochloric, hydrobromic, sulfuric, nitric or phosphoric acid; and organic acids e.g. succinic, maleic, malic, mandelic, acetic, fumaric, glutamic, lactic, citric, tartaric, benzoic, benzenesulfonic, p- toluenesulfonic, methanesulfonic, ethanesulfonic or naphthalenesulfonic acid. Other non- pharmaceutically acceptable salts e.g. oxalates, may be used, for example in the isolation of compounds of formula (I) and are included within the scope of this invention. Reference is made to Berge et al. J. Pharm. ScL, 1977, 66, 1-19.
Certain of the compounds of formula (I) may form acid addition salts with one or more equivalents of the acid. The present invention includes within its scope all possible stoichiometric and non-stoichiometric forms thereof.
Solvates of the compounds of formula (I) and solvates of the salts of the compounds of formula (I) are included within the scope of the present invention.
As used herein, the term "solvate" refers to a complex of variable stoichiometry formed by a solute (in this invention, a compound of formula (I) or a salt thereof) and a solvent. Such solvents for the purpose of the invention may not interfere with the biological activity of the solute. Examples of suitable solvents include, but are not limited to, water, methanol, ethanol and acetic acid. Preferably the solvent used is a pharmaceutically acceptable solvent. Most preferably the solvent used is water and the solvate may also be referred to as a hydrate.
Solvates of compounds of formula (I) which are suitable for use in medicine are those wherein the solvent is pharmaceutically acceptable. However, solvates having non- pharmaceutically acceptable solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts.
Prodrugs of the compounds of formula (I) are included within the scope of the present invention.
As used herein, the term "prodrug" means a compound which is converted within the body, e.g. by hydrolysis in the blood, into its active form that has medical effects. Pharmaceutically acceptable prodrugs are described in T. Higuchi and V. Stella, Prodrugs as Novel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series, and in Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987 and in D. Fleishner, S. Ramon and H. Barba "Improved oral drug delivery: solubility limitations overcome by the use of prodrugs", Advanced Drug Delivery Reviews (1996) 19(2) 1 15-130. Prodrugs are any covalently bonded carriers that release a compound of structure (I) in vivo when such prodrug is administered to a patient. Prodrugs are generally prepared by modifying functional groups in a way such that the modification is cleaved in vivo yielding the parent compound. Prodrugs may include, for example, compounds of this invention wherein hydroxy or amine groups are bonded to any group that, when administered to a patient, cleaves to form the hydroxy or amine groups. Thus, representative examples of prodrugs include (but are not limited to) phosphonate, carbamate, acetate, formate and benzoate derivatives of hydroxy and amine functional groups of the compounds of formula (I).
Phosphonates and carbamates may be active in their own right and/or be hydrolysable under in vivo conditions in the human body. Suitable pharmaceutically acceptable in vivo hydrolysable ester groups include those which break down readily in the human body to leave the parent acid or its salt. A phosphonate is formed by reaction with phosphorous (phosphonic) acid, by methods well known in the art. For example, phosphonates may be derivatives such as RP(O)(OR)2 and the like. A carbamate is an ester of carbamic acid.
In one aspect of the invention there is provided a compound, or a salt thereof, wherein the compound is selected from the group consisting of:
1-(2-Biphenylylmethyl)-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-4- carboxamide,
1 -[(4-Fluorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H- 1 ,2,3-triazole-
4-carboxamide, 1 -[(4-Bromophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-
4-carboxamide,
5-Methyl-1 -[(4-methylphenyl)methyl]-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-
4-carboxamide,
1-[(2-Chlorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-
4-carboxamide,
1-[(4-Chlorophenyl)methyl]-5-methyl-/V-(1 !2!3!4-tetrahydro-6-isoquinolinyl)-1 H-1 !2!3-triazole-
4-carboxamide, 1-[(3!4-Dichlorophenyl)methyl]-5-methyl-Λ/-(1 !2!3!4-tetrahydro-6-isoquinolinyl)-1 H-1 !2!3- triazole-4-carboxamide,
5-Methyl-1-(1-naphthalenylmethyl)-Λ/-(1 !2!3!4-tetrahydro-6-isoquinolinyl)-1 H-1 !2!3-triazole-4- carboxamide,
1 -[(3-Chlorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole- 4-carboxamide,
1-{[(2-Chlorophenyl)oxy]methyl}-5-methyl-Λ/-(1 !2!3!4-tetrahydro-6-isoquinolinyl)-1 H-1 !2!3- triazole-4-carboxamide,
5-Methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 -{[4-(trifluoromethyl)phenyl]methyl}-1 H-1 ,2,3- triazole-4-carboxamide, 1-[(2,5-Dichlorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide,
5-Methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 -{[3-(trifluoromethyl)phenyl]methyl}-1 H-1 ,2,3- triazole-4-carboxamide,
1-[(5-Chloro-1-benzothien-3-yl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H- 1 ,2,3-triazole-4-carboxamide,
1-[(5-Chloro-2-thienyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide,
1-[(2,3-Dichlorophenyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide, 1-[(3,4-Difluorophenyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide,
1-(1-Benzothien-3-ylmethyl)-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-
4-carboxamide, i-P-Chloro^-fluorophenyl^ethyO-S-methyl-N^I ^.S^-tetrahydro-θ-isoquinolinyl^i H-I ^.S- triazole-4-carboxamide,
1-[(3,5-Dichlorophenyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide,
1-{[3-Chloro-4-(trifluoromethyl)phenyl]methyl}-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-
1 H-1 ,2,3-triazole-4-carboxamide, i-p^-DichlorophenyOmethyO-Λ/^I ^.S^-tetrahydro-θ-isoquinolinyO-I H-I ^.S-triazole^- carboxamide,
1-(1-Naphthalenylmethyl)-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1H-1 ,2,3-triazole-4- carboxamide,
1 -[(3,4-Dichlorophenyl)methyl]-5-ethyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide, and
N-{1-[(3,4-Dichlorophenyl)methyl]-5-methyl-1 H-1 ,2,3-triazol-4-yl}-1 ,2,3,4-tetrahydro-6- isoquinolinecarboxamide.
In one aspect of the invention there is provided a compound selected from the group consisting of:
1-(2-Biphenylylmethyl)-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-4- carboxamide hydrochloride, 1 -[(4-Fluorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-
4-carboxamide hydrochloride,
1-[(4-Bromophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-
4-carboxamide hydrochloride,
5-Methyl-1 -[(4-methylphenyl)methyl]-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole- 4-carboxamide hydrochloride,
1-[(2-Chlorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-
4-carboxamide hydrochloride,
1-[(4-Chlorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-
4-carboxamide hydrochloride, 1-[(3,4-Dichlorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide hydrochloride,
5-Methyl-1-(1-naphthalenylmethyl)-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-4- carboxamide hydrochloride,
1 -[(3-Chlorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole- 4-carboxamide hydrochloride,
1-{[(2-Chlorophenyl)oxy]methyl}-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide hydrochloride,
5-Methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 -{[4-(trifluoromethyl)phenyl]methyl}-1 H-1 ,2,3- triazole-4-carboxamide, 1 -[(2,5-Dichlorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide,
5-Methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 -{[3-(trifluoromethyl)phenyl]methyl}-1 H-1 ,2,3- triazole-4-carboxamide, i-^S-Chloro-i-benzothien-S-yOmethyO-S-methyl-N^I ^.S^-tetrahydro-θ-isoquinolinyO-I H- 1 ,2,3-triazole-4-carboxamide hydrochloride,
1-[(5-Chloro-2-thienyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide hydrochloride,
1-[(2,3-Dichlorophenyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide hydrochloride, 1-[(3,4-Difluorophenyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide hydrochloride,
1-(1-Benzothien-3-ylmethyl)-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-
4-carboxamide hydrochloride, i-P-Chloro^-fluorophenyOmethyO-δ-methyl-N^I ^.S^-tetrahydro-θ-isoquinolinyO-I H-I ^.S- triazole-4-carboxamide hydrochloride,
1-[(3,5-Dichlorophenyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide hydrochloride,
1-{[3-Chloro-4-(trifluoromethyl)phenyl]methyl}-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)- 1 H-1 ,2,3-triazole-4-carboxamide hydrochloride,
1-[(3,4-Dichlorophenyl)methyl]-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-4- carboxamide hydrochloride, 1-(1-Naphthalenylmethyl)-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1H-1 ,2,3-triazole-4- carboxamide hydrochloride,
1-[(3,4-Dichlorophenyl)methyl]-5-ethyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide hydrochloride and
N-{1-[(3,4-Dichlorophenyl)methyl]-5-methyl-1 H-1 ,2,3-triazol-4-yl}-1 ,2,3,4-tetrahydro-6- isoquinolinecarboxamide hydrochloride.
The compounds of the invention have been found to inhibit SCD activity and may therefore be useful in regulating lipid levels, e.g. plasma lipid levels. Diseases or conditions caused by or associated with an abnormal plasma lipid profile include dyslipidemia, hypoalphalipoproteinemia, hyperbetalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial hypercholesterolemia, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, atherosclerosis, obesity, Type I diabetes, Type Il diabetes, insulin resistance, hyperinsulinaemia and metabolic syndrome. Other cardiovascular diseases for which the compounds of the present invention are useful include peripheral vascular disease, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes and thrombosis. Other diseases or conditions include hepatic steatosis, non-alcoholic steatohepatitis (NASH) and other diseases related to accumulation of lipids in the liver.
The compounds of the invention may also be useful in the treatment of skin disorders e.g. eczema, acne, psoriasis, keloid scar formation or prevention, and diseases related to production or secretions from mucous membranes.
The compounds of the invention may also be useful in the treatment of cancer, neoplasia, malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like.
The compounds of the invention may also be useful in the treatment of mild cognitive impairment (MCI), Alzheimer's disease (AD), cerebral amyloid angiopathy (CAA) or dementia associated with Down Syndrome (DS) and other neurodegenerative diseases characterized by the formation or accumulation of amyloid plaques comprising Aβ42.
Within the context of the present invention, the terms describing the indications used herein are classified in the Merck Manual of Diagnosis and Therapy, 17th Edition and/or the International Classification of Diseases 10th Edition (ICD-10). The various subtypes of the disorders mentioned herein are contemplated as part of the present invention.
In one aspect, the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in medical therapy.
In one aspect, the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating and/or preventing a disease or a condition susceptible to amelioration by an SCD inhibitor.
In another aspect, the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating and/or preventing acne, cancer, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or nonalcoholic steatohepatitis (NASH).
In another aspect, the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating and/or preventing acne, cancer, dyslipidemia, atherosclerosis, insulin resistance, hyperinsulinaemia, Type Il diabetes and/or hepatic steatosis.
In another aspect, the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating and/or preventing acne.
In one aspect, the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in treating and/or preventing a disease or a condition susceptible to amelioration by an SCD inhibitor in a mammal, including human.
In another aspect, the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in treating and/or preventing acne, cancer, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or non-alcoholic steatohepatitis (NASH).
In another aspect, the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in treating and/or preventing acne, cancer, dyslipidemia, atherosclerosis, insulin resistance, hyperinsulinaemia, Type Il diabetes and/or hepatic steatosis.
In another aspect, the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in treating and/or preventing acne.
In one aspect, the invention provides a method for treating and/or preventing a disease or a condition susceptible to amelioration by an SCD inhibitor, which method comprises administering to a subject, for example a mammal, including human, a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
In another aspect, the invention provides a method for treating and/or preventing a acne, cancer, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or non-alcoholic steatohepatitis (NASH), which method comprises administering to a subject, for example a mammal, including human, a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
In another aspect, the invention provides a method for treating and/or preventing acne, cancer, dyslipidemia, atherosclerosis, insulin resistance, hyperinsulinaemia, Type Il diabetes and/or hepatic steatosis, which method comprises administering to a subject, for example a mammal, including human, a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
In another aspect, the invention provides a method for treating and/or preventing acne, which method comprises administering to a subject, for example a mammal, including human, a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
It will be appreciated that reference to "treatment" and "therapy" includes acute treatment or prophylaxis as well as the alleviation of established symptoms.
Since the compounds of the invention are intended for use in pharmaceutical compositions it will readily be understood that they are each preferably provided in substantially pure form, for example at least 60% pure, more suitably at least 75% pure and preferably at least 85%, especially at least 98% pure (% are on a weight for weight basis). Impure preparations of the compounds may be used for preparing the more pure forms used in the pharmaceutical compositions; these less pure preparations of the compounds should contain at least 1%, more suitably at least 5% and preferably from 10 to 59% of a compound of the invention.
Processes for the preparation of the compounds of formula (I) form further aspects of the invention. R1, R2, R3 and R4 are as defined above unless otherwise specified. Throughout the specification, general formulae are designated by Roman numerals (I), (II), (III), (IV) etc.
In certain instances final compounds of formula (I) can be converted into other compounds of formula (I) by techniques known to those in the art, for example, carboxylic acid substituents can be converted to esters or amides by routine techniques.
In a general process, compounds of formula (I), wherein X represents -CONH- (formula (Ia)) may be prepared according to reaction scheme 1 by reacting compounds of formula (III) and compounds of formula (IV) wherein P1 represents a suitable nitrogen protecting group such as Boc to form a compound of formula (II). The reaction is suitably carried out in the presence of a coupling reagent such as HATU and a base such as DIPEA in a suitable solvent such as DCM or DMF (suitably at room temperature to 800C), and is followed by
deprotection of compound of formula (II) under acidic conditions such as hydrochloric acid in a suitable solvent such as ethyl acetate (suitably at room temperature).
Scheme 1

Accordingly, in one aspect the invention provides a process for the preparation of compounds of the formula (Ia) by reacting compounds of formula (III), wherein R1 and R3 are defined above, with compounds of formula (IV), wherein P1 is defined above, in the presence of a coupling agent, followed by deprotection of compounds of formula (II).
Compounds of formula (III) may be prepared according to reaction scheme 2 by reacting compounds of formula (V) and compounds of formula (Vl) in the presence of base such as potassium carbonate in a suitable solvent such as DMF or DMSO (suitably at 400C to 8O0C), followed by saponification of compounds of formula (VII) in basic conditions such as sodium hydroxide in a suitable solvent, such as ethanol or methanol to reflux.
Scheme 2
Saponification

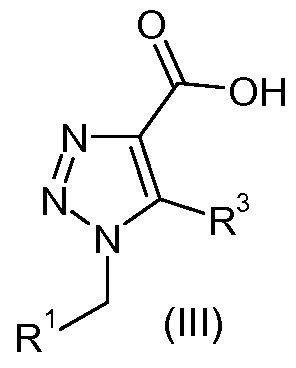
Compounds of formula (III), wherein R3 represents H (formula (NIa)) may be prepared according to reaction scheme 3 by reacting compounds of formula (V) and ethyl 2- propynoate (VIII) in a suitable solvent such as ethanol at reflux, followed by saponification of
compounds of formula (IX) in basic conditions such as sodium hydroxide in a suitable solvent, such as ethanol or methanol to reflux.
Scheme 3

In a general process, compounds of formula (I) wherein X represents -NHCO- (formula (Ib)) may be prepared according to reaction scheme 4 by reacting compounds of formula (Xl) and compound of formula (XII) wherein P1 represents a suitable nitrogen protecting group such as Boc to form a compound of formula (X). The reaction is suitably carried out in the presence of a coupling reagent such as HATU and a base such as DIPEA in a suitable solvent such as DCM or DMF (suitably at room temperature to 800C), and is followed by deprotection of compound of formula (X) under acidic conditions such as hydrochloric acid in ethyl acetate (suitably at room temperature).
Scheme 4

Accordingly, in one aspect the invention provides a process for the preparation of compounds of the formula (Ib) by reacting compounds of formula (Xl), wherein R1 and R3 are defined above, with compounds of formula (XII), wherein wherein P1 is defined above, in the presence of a coupling agent, followed by deprotection of compounds of formula (X).
Compounds of formula (Xl) may be prepared according to reaction scheme 5 by reacting compounds of formula (XIa) in the presence of bromine and a base such as potassium hydroxide in a suitable solvent such as water (suitably at 4O0C to 800C).
Scheme 5

Compounds of formula (XIa) may be prepared according to reaction scheme 6 starting from compounds of formula (III) in the presence of thionyl chloride in chloroform at room temperature followed by reaction with aqueous ammonia in acetonitrile on ice (e.g at -5°C to 50C).
Scheme 6

Compounds of formula (IV), (V), (Vl), (VIII) and (XII) are commercially available or may be prepared by methods known in the literature or processes known to those skilled in the art.
Further details for the preparation of compounds of formula (I) are found in the examples section hereinafter.
The compounds of the invention may be prepared singly or as compound libraries comprising at least 2, for example 5 to 1 ,000 compounds, and more preferably 10 to 100 compounds. Libraries of compounds of the invention may be prepared by a combinatorial
'split and mix' approach or by multiple parallel syntheses using either solution phase or solid phase chemistry, by procedures known to those skilled in the art. Thus according to a further aspect there is provided a compound library comprising at least 2 compounds of the invention.
Those skilled in the art will appreciate that in the preparation of compounds of formula (I) and/or salts thereof it may be necessary and/or desirable to protect one or more sensitive groups in the molecule or the appropriate intermediate to prevent undesirable side reactions. Suitable protecting groups for use according to the present invention are well known to those
skilled in the art and may be used in a conventional manner. See, for example, "Protective groups in organic synthesis" by T.W. Greene and P. G. M. Wuts (John Wiley & sons 1991 ) or "Protecting Groups" by PJ. Kocienski (Georg Thieme Verlag 1994). Examples of suitable amino protecting groups include acyl type protecting groups (e.g. formyl, trifluoroacetyl, acetyl), aromatic urethane type protecting groups (e.g. benzyloxycarbonyl (Cbz) and substituted Cbz), aliphatic urethane protecting groups (e.g. 9-fluorenylmethoxycarbonyl (Fmoc), t-butyloxycarbonyl (Boc), isopropyloxycarbonyl, cyclohexyloxycarbonyl) and alkyl or aralkyl type protecting groups (e.g. benzyl, trityl, chlorotrityl).
Various intermediate compounds used in the above-mentioned process, including but not limited to certain compounds of formulae (II) and (X), constitute a further aspect of the present invention.
The compounds of formula (I) or pharmaceutically acceptable salt(s) thereof may also be used in combination with other therapeutic agents. The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or pharmaceutically acceptable salt thereof together with one or more further therapeutic agent(s).
Compounds of the invention may be administered in combination with other therapeutic agents. Preferred therapeutic agents are selected from the list: an inhibitor of cholesteryl ester transferase (CETP inhibitors), a HMG-CoA reductase inhibitor, a microsomal triglyceride transfer protein, a peroxisome proliferator-activated receptor activator (PPAR), a bile acid reuptake inhibitor, a cholesterol absorption inhibitor, a cholesterol synthesis inhibitor, a fibrate, niacin, an ion-exchange resin, an antioxidant, an inhibitor of AcylCoA: cholesterol acyltransferase (ACAT inhibitor), a cannabinoid 1 antagonist, a bile acid sequestrant, a corticosteroid, a vitamin D3 derivative, a retinoid, an immunomodulator, an anti androgen, a keratolytic agent, an anti-microbial, a platinum chemotherapeutic, an antimetabolite, hydroxyurea, a taxane, a mitotic disrupter, an anthracycline, dactinomycin, an alkylating agent and a cholinesterase inhibitor.
When the compound of formula (I) or pharmaceutically acceptable salt thereof is used in combination with a second therapeutic agent the dose of each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art. It will be appreciated that the amount of a compound of the invention required for use in treatment will vary with the nature of the condition being treated and the age and the condition of the patient and will be ultimately at the discretion of the attendant physician or veterinarian.
The combinations referred to above may conveniently be presented for use in the form of a pharmaceutical formulation and thus pharmaceutical formulations comprising a combination as defined above together with at least one pharmaceutically acceptable carrier and/or excipient comprise a further aspect of the invention. The individual components of such
combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations by any convenient route.
When administration is sequential, either the SCD inhibitor or the second therapeutic agent may be administered first. When administration is simultaneous, the combination may be administered either in the same or different pharmaceutical composition.
When combined in the same formulation it will be appreciated that the two compounds must be stable and compatible with each other and the other components of the formulation. When formulated separately they may be provided in any convenient formulation, conveniently in such manner as are known for such compounds in the art.
The invention also includes a pharmaceutical composition comprising one or more compounds of formula (I) or pharmaceutically acceptable salt (s) in combination with one or more excipients.
The compounds of the invention may be administered in conventional dosage forms prepared by combining a compound of the invention with standard pharmaceutical carriers or diluents according to conventional procedures well known in the art. These procedures may involve mixing, granulating and compressing or dissolving the ingredients as appropriate to the desired preparation.
The pharmaceutical compositions of the invention may be formulated for administration by any route, and include those in a form adapted for oral, topical or parenteral administration to mammals including humans.
The compositions may be in the form of tablets, capsules, powders, granules, lozenges, creams or liquid preparations, such as oral or sterile parenteral solutions or suspensions.
The topical formulations of the present invention may be presented as, for instance, dispersions, lotions, creams, gels, pastes, powders, aerosol sprays, syrups or ointments on sponges or cotton applicators, and solutions or suspensions in an aqueous liquid, nonaqueous liquid, oil-in-water emulsion, or water-in-oil liquid emulsion.
Creams, lotions, or ointments, may be prepared as rinse-off or leave-on products, as well as two stage treatment products for use with other skin cleansing or managing compositions. The compositions can be administered as a rinse-off product in a higher concentration form, such as a gel, and then a leave-on product in a lower concentration to avoid irritation of the skin. Each of these forms is well understood by those of ordinary skill in the art, such that dosages may be easily prepared to incorporate the pharmaceutical composition of the invention.
Ointments are hydrocarbon-based semisolid formulations containing dissolved or suspended drugs. Creams and lotions are semi-solid emulsion systems and the term is applied both to water/oil or oil/water. Gel formulations are semi-solid systems in which a liquid phase is trapped in a polymeric matrix.
By way of non-limiting example, the ointments may contain one or more hydrophobic carriers selected from, for example, white soft paraffin or other mineral waxes, liquid paraffin, non- mineral waxes, long chain alcohols, long chain acids and silicones. The ointment may contain in addition to the hydrophobic carriers some hydrophillic carriers selected from, for example, propylene glycol and polyethylene glycol in combination with an appropriate surfactant/co-surfactant system. The carrier compositions of the creams or lotions are typically based on water, white soft paraffin and an appropriate surfactant/co-surfactant system, in combination with other carriers/components selected from, for example, propylene glycol, butylene glycol glycerinemonostearate, PEG-glycerinemonostearate, esters such as Ci2-i5 alkyl benzoate, liquid paraffin, non-mineral waxes, long chain alcohols, long chain acids silicones, non-silicone polymers. The gels may by way of example be formulated using isopropyl alcohol or ethyl alcohol, propylene glycol and water with a gelling agent such as hydroxyethyl cellulose, suitably in combination with minor components, for example one or more of butylene glycol and a wetting agent such as a poloxamer.
An ointment, cream, lotion, gel, and the like, can further comprise a moisturizing agent. The moisturizing agent can be a hydrophobic moisturizing agent such as ceramide, borage oil, tocopherol, tocopherol linoleate, dimethicone or a mixture thereof or a hydrophilic moisturizing agent such as glycerine, hyaluronic acid, sodium peroxylinecarbolic acid, wheat protein, hair keratin amino acids, or a mixture thereof.
The compositions according to the invention may also comprise conventional additives and adjuvants for dermatological applications, such as preservatives, acids or bases used as pH buffer excipients and antioxidants.
The present invention encompasses administration via a transdermal patch or other forms of transdermal administration. Suitable formulations for transdermal administration are known in the art, and may be employed in the methods of the present invention. For example, suitable transdermal patch formulations for the administration of a pharmaceutical compound are described in, for example, U.S. Pat. No. 4, 460,372 to Campbell et al., U.S. Pat. No. 4,573,996 to Kwiatek et al., U. S. Pat. No. 4,624,665 to Nuwayser, U.S. Pat. No. 4,722,941 to Eckert et al., and U.S. Pat. No. 5, 223,261 to Nelson et al.
One suitable type of transdermal patch for use in the methods of the present invention encompasses a suitable transdermal patch includes a backing layer which is non-permeable, a permeable surface layer, an adhesive layer substantially continuously coating the permeable surface layer, and a reservoir located or sandwiched between the backing layer and the permeable surface layer such that the backing layer extends around the sides of the
reservoir and is joined to the permeable surface layer at the edges of the permeable surface layer. The reservoir contains a compound of formula (I) or pharmaceutically acceptable salt thereof, alone or in combination, and is in fluid contact with the permeable surface layer. The transdermal patch is adhered to the skin by the adhesive layer on the permeable surface layer, such that the permeable surface layer is in substantially continuous contact with the skin when the transdermal patch is adhered to the skin. While the transdermal patch is adhered to the skin of the subject, the compound of formula (I) or pharmaceutically acceptable salt thereof contained in the reservoir of the transdermal patch is transferred via the permeable surface layer, from the reservoir, through the adhesive layer, and to the skin of the patient. The transdermal patch may optionally also include one or more penetration- enhancing agents in the reservoir that enhance the penetration of the compound of formula (I) or pharmaceutically acceptable salt thereof through the skin.
Examples of suitable materials which may comprise the backing layer are well known in the art of transdermal patch delivery, and any conventional backing layer material may be employed in the transdermal patch of the instant invention.
Suitable penetration-enhancing agents are well known in the art as well. Examples of conventional penetration-enhancing agents include alkanols such as ethanol, hexanol, cyclohexanol, and the like, hydrocarbons such as hexane, cyclohexaue, isopropylbenzene; aldebydes and ketones such as cyclohexanone, acetamide, N,N-di(lower alkyl)acetamides such as N,N-diethylacetamide, N,N-dimethyl acetamide, N-(2-hydroxyethyl) acetamide, esters such as N,N-di-lower alkyl sulfoxides; essential oils such as propylene glycol, glycerine, glycerol monolaurate, isopropyl myristate, and ethyl oleate, salicylates, and mixtures of any of the above.
Tablets and capsules for oral administration may be in unit dose presentation form, and may contain conventional excipients such as binding agents, for example syrup, acacia, gelatin, sorbitol, tragacanth, or polyvinylpyrrolidone; fillers, for example lactose, sugar, maize-starch, calcium phosphate, sorbitol or glycine; tabletting lubricants, for example magnesium stearate, talc, polyethylene glycol or silica; disintegrants, for example potato starch; or acceptable wetting agents such as sodium lauryl sulphate. The tablets may be coated according to methods well known in normal pharmaceutical practice. Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use. Such liquid preparations may contain conventional additives, such as suspending agents, for example sorbitol, methyl cellulose, glucose syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminium stearate gel or hydrogenated edible fats, emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which may include edible oils), for example almond oil, oily esters such as glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and, if desired, conventional flavouring or colouring agents.
Preparations for oral administration may be suitably formulated to give controlled/extended release of the active compound.
Suppositories will contain conventional suppository bases, e.g. cocoa-butter or other glyceride.
For parenteral administration, fluid unit dosage forms are prepared utilising the compound and a sterile vehicle, water being preferred. The compound, depending on the vehicle and concentration used, can be either suspended or dissolved in the vehicle. In preparing solutions the compound can be dissolved in water for injection and filter sterilised before filling into a suitable vial or ampoule and sealing.
Advantageously, agents such as a local anaesthetic, preservative and buffering agents can be dissolved in the vehicle. To enhance the stability, the composition can be frozen after filling into the vial and the water removed under vacuum. The dry lyophilised powder is then sealed in the vial and an accompanying vial of water for injection may be supplied to reconstitute the liquid prior to use. Parenteral suspensions are prepared in substantially the same manner except that the compound is suspended in the vehicle instead of being dissolved and sterilisation cannot be accomplished by filtration. The compound can be sterilised by exposure to ethylene oxide before suspending in the sterile vehicle. Advantageously, a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
The compositions may contain from 0.1 % by weight, preferably from 10-60% by weight, of the active ingredient, depending on the method of administration. Where the compositions comprise dosage units, each unit will preferably contain from 50-500 mg of the active ingredient. The dosage as employed for adult human treatment will preferably range from
100 to 3000 mg per day, for instance 1500 mg per day depending on the route and frequency of administration. Such a dosage corresponds to 1.5 to 50 mg/kg per day. Suitably the dosage is from 5 to 20 mg/kg per day.
It will be recognised by one of skill in the art that the optimal quantity and spacing of individual dosages of a compound of the invention will be determined by the nature and extent of the condition being treated, the form, route and site of administration, and the particular mammal being treated, and that such optimums can be determined by conventional techniques. It will also be appreciated by one of skill in the art that the optimal course of treatment, i.e., the number of doses of a compound of the invention given per day for a defined number of days, can be ascertained by those skilled in the art using conventional course of treatment determination tests.
The invention also extends to novel intermediates disclosed herein, used in the preparation of compounds of formula (I) or salts thereof.
DEFINITIONS
Boc tertbutyloxy carbonyl
DIPEA diisopropylethylamine
DCM dichloromethane DMF dimethylformamide
DMSO dimethyl sulfoxide
EtOAc ethyl acetate
HATU O-(7-Azabenzotriazol-1-yl)-1 ,1 ,3,3-tetramethyluronium hexafluorophosphate HCI hydrochloric acid
MeOH methanol
NaOH sodium hydroxide
Regardless of how the preparation of compounds are represented in the present specification no inference can be drawn that particular batches (or mixtures of two or more batches) of intermediates were used in the next stage of the preparation. The examples and intermediates are intended to illustrate the synthetic routes suitable for preparation of the same, to assist the skilled persons understanding of the present invention.
Where reference is made to the use of a "similar" procedure, as will be appreciated by those skilled in the art, such a procedure may involve minor variation, for example reaction temperature, reagent/solvent amount, reaction time, work-up conditions or chromatographic purification conditions.
Analytical methods LC-MS
Analytical HPLC was conducted on a X-terra MS C18 column (2.5 μm 3 x 30 mm id) eluting with 0.01 M ammonium acetate in water (solvent A) and 100% acetonitrile using the following elution gradient: 0 to 4 minutes, 5 to 100%B; 4 to 5 minutes, 100%B at a flow-rate of 1.1 mL/min with a temperature of 400C.
The mass spectra (MS) were recorded on a micromass ZQ-LC mass spectrometer using electrospray positive ionisation [ES+ve to give MH+ molecular ion] or electrospray negative ionisation [ES-ve to give (M-H)" molecular ion] modes.
Analytical methods LC-HRMS
Analytical HPLC was conducted on an Uptisphere-hsc column (3 μm 30 x 3 mm id) eluting with 0,01 M ammonium acetate in water (solvent A) and 100% acetonitrile (solvent B) using the following elution gradient: 0 to 0.5 minutes, 5%B; 0.5 to 3.5 minutes, 5 to 100%B; 3.5 to 4 minutes, 100%B; 4 to 4.5 minutes, 100 to 5%B; 4.5 to 5.5 minutes, 5%B at a flow-rate of 1.3 mL/min with a temperature of 400C.
The mass spectra (MS) were recorded on a micromass LCT, mass spectrometer using electrospray positive ionisation [ES+ve to give MH+ molecular ion] or electrospray negative ionisation [ES-ve to give (M-H)" molecular ion] modes.
Analytical method GC-MS
Analytical GC was conducted on a DB-1 ms column (Agilent Technologies), 0.1 μm 10m x 0.1 mm id) eluting with an Helium flow of 0.5ml/min and pressure at 3.4 bar and with a gradient temperature: 0 to 0.35 min, 1000C; 0.35min to 6min, 1000C to 250°C (ramp of 80°C/min).
The mass spectra (MS) were recorded on a Agilent Technologies G5973 mass spectrometer using electronic impact ionisation.
The following non-limiting examples illustrate the present invention.
Intermediate 1 : Sodium bromomethanesulfonate
Br^^SO3 " Na+
A mixture of dibromomethane (93g, 0.52mol) and sodium sulfite (65.25g, 0.52mol) in water (19OmL) was stirred and heated under reflux for 80 hours and then 15OmL of water was distilled. The solid that separated was filtered and recrystallized from water to give the title compound (9Og, 88%).
Intermediate 2: [(2-Chlorophenyl)oxy1methanesulfonic acid

To a solution of sodium hydroxide (10.25g, 0.26mol) in 35mL of water 2-chlorophenol (30.75g, 0.24mol) was added. The mixture was heated and stirred at 800C for 1 hour. Sodium bromomethanesulfonate (Intermediate 1 ) (45g, 0.23mol) was added. The mixture was stirred at 800C for 1 hour, and then water was evaporated. The solid residue was heated at 200°C for 25 hours. After it was cooled to room temperature, the solid was dissolved in 40OmL of warm water, filtered, and the filtrate was adjusted to pH=5. The solution was cooled, washed with two 10OmL portions of diethyl ether, and then concentrated to 10OmL in vacuum. The cooled solution led to a crystalline solid which was filtered to give the title compound as a mixture of acid and sodium salt (18g, 35%). 1HNMR (300MHz, CDCI3) δ: 7.38-6.92 (m, 4H), 4.62 (s, 2H).
Intermediate 3: Chloromethyl 2-chlorophenyl ether

A mixture of [(2-Chlorophenyl)oxy]methanesulfonic acid (Intermediate 2) (13.Og, 0.058mol) and phosphorus pentachloride (28.9g, 0.14mol) in diethyl ether was stirred at room temperature for 2 hours. The mixture was diluted with 40OmL of diethyl ether and the solution poured on 40Og of crushed ice. The organic phase was separated and the water phase was extracted with diethyl ether twice. The organic phases were combined, washed with brine, dried over Na2SO4, and concentrated to give the title compound as a red oil (8.4g, 81%). 1HNMR (300MHz, CDCI3) δ: 7.40-7.25(m, 4H), 5.92 (s, 2H).
Intermediate 4: 4-(Bromomethyl)-2-chloro-1-(trifluoromethyl)benzene

To a solution of 2-chloro-4-methyl-1-(trifluoromethyl)benzene (0.5g, 2.57 mmol) in dry carbone tetrachloride (5OmL), N-bromo succinimide (0.412g, 2.313mmol) and benzoyl peroxide (12mg, 0.05 mmol) were added. The reaction was stirred to reflux for 40 hours. After cooling at 00C, the reaction mixture was filtered and the solvent evaporated. The title compound was obtained (0.7g) and the crude compound was used in the next reaction without purification. GC/MS: m/z, 273 Rt: 5.5 min
The following Intermediates were prepared using the generic reaction scheme (Scheme 1 )
R!
Br R1
R1
N,
Cl
Intermediate 5: 2-Biphenylylmethyl azide

The following Intermediates were similarly prepared by a method analogous to that described for Intermediate 5:
1
R
N,
Table 1



The following Intermediates were prepared using the generic reaction scheme (Scheme 2)

Intermediate 24: Methyl 1-(2-biphenylylmethyl)-5-methyl-1 /-/-1 ,2,3-triazole-4-carboxylate

To a solution of 2-biphenylylmethyl azide (Intermediate 5) (1.1 g, 5.2mmol) and methyl acetoacetate (0.9g, 7.8mmol) in DMSO (1 OmL), K2CO3 (2.9g, 20mmol) was added. The reaction mixture was stirred at 400C for 48 hours. After cooling, water was added and the mixture was extracted with ethyl acetate, the organic layer was dried on sodium sulfate and after filtration was evaporated to dryness. The residue was purified by flash column chromatography eluting with DCM and DCM/MeOH: 98/2 to give the title compound as a colourless oil (1.1g, 69%). LC/MS: m/z 308 (M+H)+, Rt: 3.14 min.
The following Intermediates were similarly prepared by a method analogous to that described for Intermediate 24:

Table 2



Intermediate 43: Ethyl 1-[(3,4-dichlorophenyl)methyl1-1H-1,2,3-triazole-4-carboxylate

To a solution of (3,4-dichlorophenyl)methyl azide (Intermediate 9) (10g, 53.2mmol) in ethanol (30OmL), ethyl propiolate (5.2g, 53mmol) was added. The mixture was heated to reflux for 5 days. The solvent was removed under reduced pressure. The residue was purified by flash column chromatography eluting with pentane/EtOAc (4:1 to 1 :2) to give the title compound (11.1 g, 66%). LC/MS: m/z 300 (M+H)+, Rt: 2.78 min.
The following Intermediate was similarly prepared by a method analogous to that described for Intermediate 43:

Table 3

Intermediate 45: Methyl 1-[(3,4-dichlorophenyl)methyl1-5-ethyl-1 H-1 ,2,3-triazole-4- carboxylate

To a solution of (3,4-dichlorophenyl)methyl azide (Intermediate 9) (1 1.6g, 57.6mmol) in DMSO (4OmL), methyl propionylacetate (5g, 38.4mmol) and potassium carbonate (21.2g, 0.154mol) were added. The mixture was heated at 45°C for 2 days. The reaction was cooled to room temperature and then poured into ice-water. The yellow solid was filtered and dried
in vacuum. The solid was then washed with ethyl acetate, filtered and dried to give the title compound as a white solid (9.5 g, 78%). LC/MS: m/z 314 (IVH-H)+, Rt: 2.76 min.
The following Intermediates were prepared using the generic reaction scheme (Scheme 3)

Intermediate 46: 1-(2-Biphenylylmethyl)-5-methyl-1 H-1 ,2,3-triazole-4-carboxylic acid

To a solution of methyl 1-(2-biphenylylmethyl)-5-methyl-1 H-1 ,2,3-triazole-4-carboxylate (Intermediate 24) (1.1g, 3.7mmol.) in methyl alcohol, an aqueous solution of sodium hydroxide (7.2ml, 6 mmol) was added. The solution was heated at reflux for 3 hours. After concentration under reduced pressure, a 1 N hydrogen chloride solution (8 ml) was added. The resulting solid material was filtered, washed with water and dried to give the title compound as a cream solid (810mg, 77%). LC/MS: m/z 294 (M+H)+, Rt: 2.23 min.
The following Intermediates were similarly prepared by a method analogous to that described for Intermediate 46:

Table 4




Intermediate 65: 1-r(3,4-Dichlorophenyl)methyll-1 H-1 ,2,3-triazole-4-carboxylic acid

To a solution of ethyl 1-[(3,4-dichlorophenyl)methyl]-1 H-1 ,2,3-triazole-4-carboxylate (Intermediate 43) (3g, lOmmol) in methanol (6OmL), a sodium hydroxide solution (2.4g, 60mmol) in water (2OmL) was added. The mixture was stirred at room temperature overnight. Water was added and the methanol was removed under reduced pressure. The residue was adjusted to pH=2 with a 2N HCI solution. The precipitate was filtered and dried to give the title compound as a white solid (2.2g, 73%).
LC/MS: m/z 272 (IVH-H)+, Rt: 2.47 min
The following Intermediates were similarly prepared by a method analogous to that described for Intermediate 65:

Table 5


Intermediate 68: 1-r(3,4-dichlorophenyl)methyll-5-methyl-1 H-1 ,2,3-triazole-4-carboxamide
Intermediate 69: 1-r(3,4-dichlorophenyl)methyll-5-methyl-1 H-1 ,2,3-triazol-4-amine

To a solution of potassium hydroxide (10.96g, 195.8mmol) in water (5OmL), cooled in an ice- salt bath, bromine (6.3g, 32.16mmol) was added. At 00C, 1-[(3,4-dichlorophenyl)methyl]-5- methyl-1 H-1 ,2,3-triazole-4-carboxamide (Intermediate 68) (9.3g, 32.63mmol) was added for 4 hours under vigorous stirring. The reaction mixture was then warmed at 800C for 2 days and stirred at room temperature for 12 hours. The resulting solid was filtered and purified by HPLC to give the title compound (3.45g, 41.14%). LC/MS: m/z 257 (M+H)+ Rt: 2.25 min
The following Intermediates were prepared using the generic reaction scheme (Scheme 4)

Intermediate 70: 1.1-Dimethylethyl 6-αri-(2-biDhenylylmethvn-5-methyl-1 H-1.2.3-triazol-4- yllcarbonyl}amino)-3,4-dihvdro-2(1 H)-isoquinolinecarboxvlate

To a solution of 1-(2-biphenylylmethyl)-5-methyl-1 H-1 ,2,3-triazole-4-carboxylic acid
(Intermediate 46) (0.3g, 1 mmol), HATU (0.78g, 2mmol) and DIPEA (0.38mL, 2 mmol) in DMF
(1OmL) the commercially available 1 ,1-dimethylethyl 6-amino-3,4-dihydro-2(1 H)- isoquinolinecarboxylate (0.25g, 1 mmol) was added. The reaction mixture was stirred at room temperature for 48 hours. The DMF was evaporated under reduced pressure and the residue was dissolved in DCM. The organic phase was then washed with water, dried over sodium sulfate, filtered and evaporated under reduced pressure. The residue was purified by flash column chromatography eluting with DCM/MeOH: 99/1 to give the title compound as a compact oil (0.51 g, 96%). LC/MS: m/z 524 (M+H)+, Rt: 4.01 min.
The following Intermediates were similarly prepared by a method analogous to that described for Intermediate 70:

Table 6





Intermediate 91 : 1 ,1-Dimethylethyl 6-rαi-r(3.4-dichlorophenyl)methyll-1 H-1 ,2,3-triazol-4- yl}carbonyl)amino1-3,4-dihvdro-2(1 /-/)-isoquinolinecarboxylate

A mixture of 1-[(3,4-dichlorophenyl)methyl]-1 H-1,2,3-triazole-4-carboxylic acid (Intermediate 65) (0.1g, 0.37mmol), 1 ,1-dimethylethyl 6-amino-3,4-dihydro-2(1 H)-isoquinolinecarboxylate (91 mg, 0.37mmol), HATU (0.182g, 0.48mmol) and DIPEA (83μL, 0.48mmol) in DMF (5mL)
was stirred at room temperature overnight. The mixture was evaporated and the residue was washed with water (2OmL) and extracted with DCM (2OmL). The organic phase was dried over Na2SO4, filtered and concentrated to give the title compound as a brown oil (1 18mg, 64
%).
LC/MS: m/z 500 (M-H)+, Rt: 3.80 min.
The following Intermediates were similarly prepared by a method analogous to that described for Intermediate 91 :

Table 7
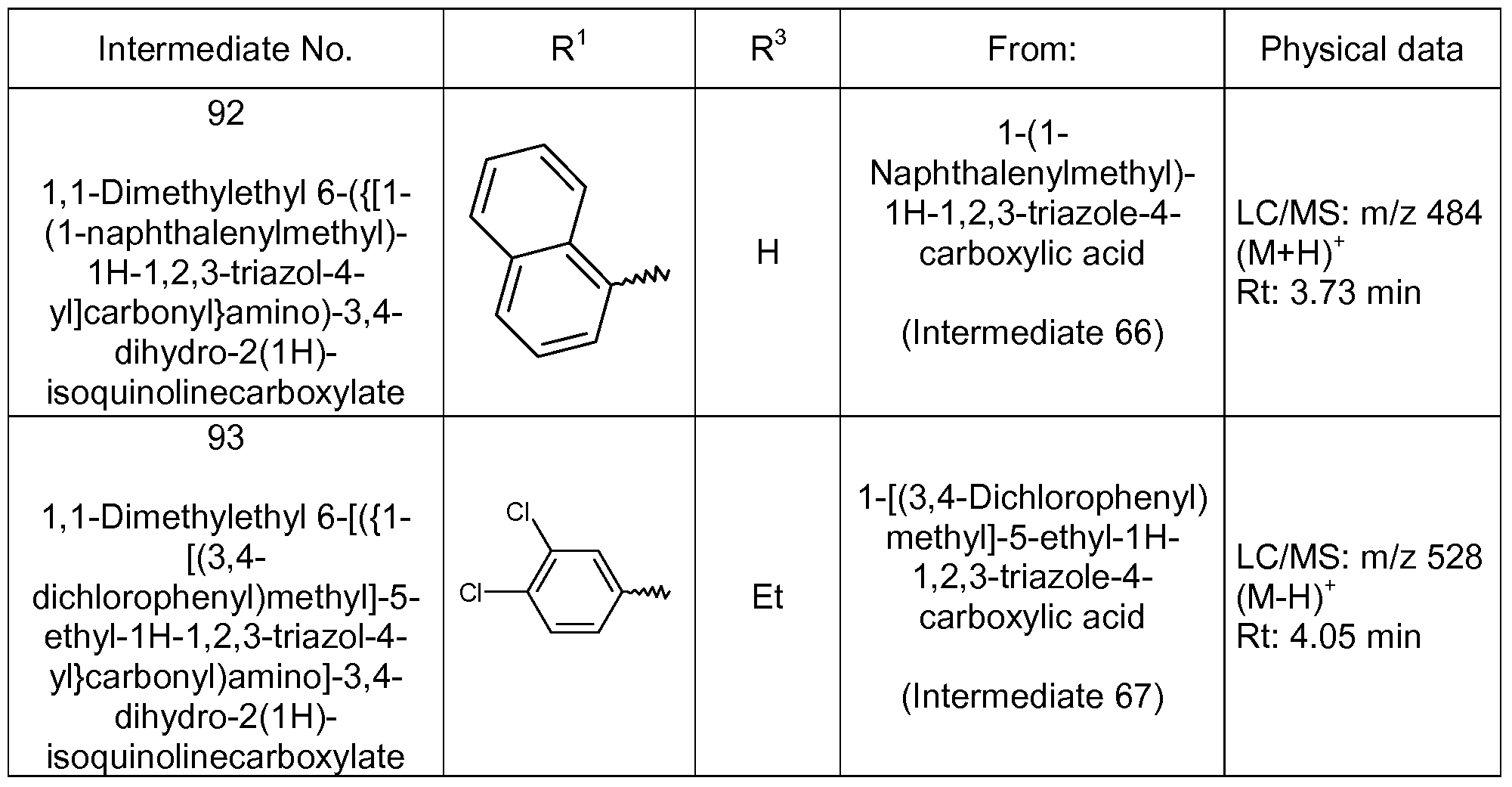
The following Intermediate was prepared using the generic reaction scheme (Scheme 6)
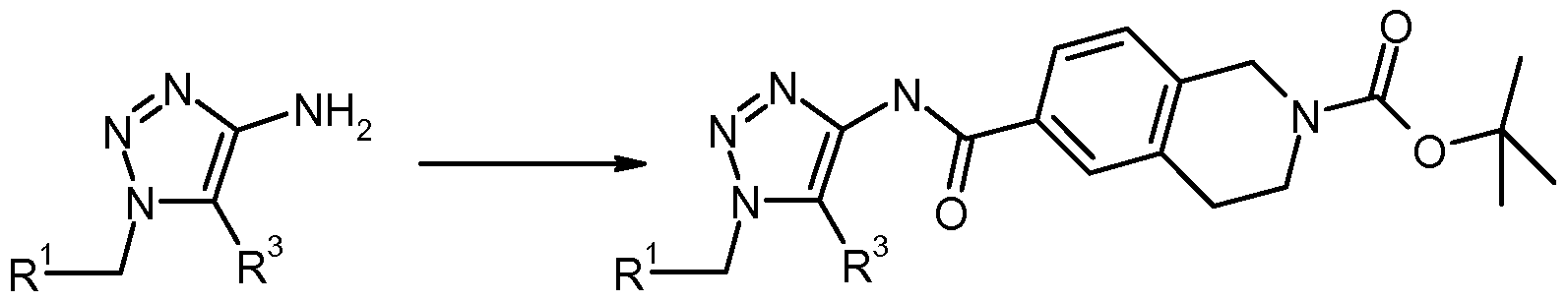
Intermediate 94: 1.1-dimethylethyl 6-r({1-r(3.4-dichlorophenyl)methyll-5-methyl-1 H-1.2.3- triazol-4-yl}amino)carbonyl1-3,4-dihvdro-2(1 H)-isoquinolinecarboxylate

A mixture of 1-[(3,4-dichlorophenyl)methyl]-5-methyl-1 H-1 ,2,3-triazol-4-amine (Intermediate 69) (0.1g, 0.39mmol), 2-{[(1 ,1-dimethylethyl)oxy]carbonyl}-1 ,2,3,4-tetrahydro-6- isoquinolinecarboxylic acid (0.1 1g, 0.37mmol), HATU (0.19g, O.δmmol) and DIPEA (88μl_, O.δmmol) in DMF (5mL) was stirred at room temperature overnight. The mixture was evaporated. The residue was washed with water and extracted with DCM. The organic phase was dried over Na2SO4, filtered and concentrated. The title compound was obtained as a white solid after purification by flash column chromatography eluting with DCM/MeOH: 98//2 and crystallisation from isopropyl ether (86mg, 43 %). HRMS calculated for C25H27CI2N5O3 (M+H)+ 516.1569, found: 516.1591 , Rt: 3.13 min.
The following Examples were prepared using the generic reaction scheme (Scheme 7)

Example 1 : 1-(2-Biphenylylmethyl)-5-methyl-Λ/-(1 ,2,3,4-tetrahvdro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide hydrochloride

HCI(g) was bubbled in EtOAc (2OmL) and a suspension of 1 ,1-dimethylethyl 6-({[1-(2- biphenylylmethyl)-5-methyl-1 H-1 ,2,3-triazol-4-yl]carbonyl}amino)-3,4-dihydro-2(1 H)- isoquinolinecarboxylate (Intermediate 70) (0.51g, 0.97 mmol) in EtOAc (1OmL) was added. The reaction mixture was stirred at room temperature for 3 hours. The resulting precipitate was filtered, washed with pentane and dried to give the title compound as a cream solid (100mg, 25%). HRMS calculated for C26H25N5O (M+H)+: 424.2137, Found: 424.2141 , Rt: 2.67 min.
MP: 1600C
The following Examples were similarly prepared by a method analogous to that described for Example 1 :

Table 8
Example No. R1 From Intermediate No: Physical data
1 ,1-Dimethylethyl 6-[({1-[(4-
HRMS (M+H)+ fluorophenyl)methyl]-5- calculated for methyl-1 H-1 ,2,3-triazol-4-
1 -[(4-Fluorophenyl)methyl]-5- C20H20FN5O yl}carbonyl)amino]-3,4- methyl-Λ/-(1 ,2,3,4-tetrahydro- Theo 366.1730, dihydro-2(1 H)-
6-isoquinolinyl)-1 H-1 ,2,3-
 Found: 366.1708, isoquinolinecarboxylate triazole-4-carboxamide Rt: 2.20 min. hydrochloride MP: 312.9°C
Found: 366.1708, isoquinolinecarboxylate triazole-4-carboxamide Rt: 2.20 min. hydrochloride MP: 312.9°C
(Intermediate 71 )
1 ,1-Dimethylethyl 6-[({1 -[(4-
HRMS (M+H)+ bromophenyl)methyl]-5- calculated for methyl-1 H-1 ,2,3-triazol-4-
1 -[(4-Bromophenyl)methyl]-5- C20H20BrN5O yl}carbonyl)amino]-3,4- methyl-Λ/-(1 ,2,3,4-tetrahydro- Theo: 426.0929 dihydro-2(1 H)-
6-isoquinolinyl)-1 H-1 ,2,3-
 Found: 426.0962 isoquinolinecarboxylate triazole-4-carboxamide Rt: 2.54 min hydrochloride MP: 315°C
Found: 426.0962 isoquinolinecarboxylate triazole-4-carboxamide Rt: 2.54 min hydrochloride MP: 315°C
(Intermediatge 72)
1 ,1-Dimethylethyl 6-[({5- methyl-1-[(4- HRMS (M+H)+ methylphenyl)methyl]-1 H- calculated for
5-Methyl-1-[(4-
1 ,2,3-triazol-4- C2i H23N5O methylphenyl)methyl]-/V- yl}carbonyl)amino]-3,4- Theo: 362.1981
(1 ,2,3,4-tetrahydro-6-
 dihydro-2(1 H)- Found: 362.2006 isoquinolinyl)-1 H-1 ,2,3- isoquinolinecarboxylate Rt: 2.45 min triazole-4-carboxamide MP: 3070C hydrochloride
dihydro-2(1 H)- Found: 362.2006 isoquinolinyl)-1 H-1 ,2,3- isoquinolinecarboxylate Rt: 2.45 min triazole-4-carboxamide MP: 3070C hydrochloride
(Intermediate 73)
1 ,1-Dimethylethyl 6-[({1-[(2- HRMS (M+H)+ chlorophenyl)methyl]-5- calculated for
1 -[(2-Chlorophenyl)methyl]-5- methyl-1 H-1 ,2,3-triazol-4- C20H20CIN5O
 methyl-Λ/-(1 ,2,3,4-tetrahydro- yl}carbonyl)amino]-3,4- Theo: 382.1435,
methyl-Λ/-(1 ,2,3,4-tetrahydro- yl}carbonyl)amino]-3,4- Theo: 382.1435,

 ,
,
 Example No. R1 From Intermediate No: Physical data
Example No. R1 From Intermediate No: Physical data
1 ,1-Dimethylethyl 6-[({1-
20 [(3,5-dichlorophenyl)methyl]- HRMS (M+H)+ 5-methyl-1 H-1 ,2,3-triazol-4- calculated for
1-[(3,5-Dichlorophenyl)methyl]- yl}carbonyl)amino]-3,4- C20H19CI2N5O
5-methyl-N-(1 , 2,3,4- dihydro-2(1 H)- Theo: 416.1045, tetrahydro-6-isoquinolinyl)-1 H-
 isoquinolinecarboxylate Found: 416.1047,
isoquinolinecarboxylate Found: 416.1047,
1 ,2,3-triazole-4-carboxamide: Rt: 2.49 min. hydrochloride (Intermediate 89) MP>260°C
1 ,1-dimethylethyl 6-{[(1-{[3-
21 chloro-4-(trifluoromethyl) HRMS (M+H)+ phenyl]methyl}-5-methyl calculated for
1-{[3-Chloro-4-
-1 H-1 ,2,3-triazol-4- C2IH19CIF3N5O
(trifluoromethyl)phenyl] yl)carbonyl]amino}-3,4- Theo: 450.1308, methyl}-5-methyl-N-(1 ,2,3,4-
 dihydro-2(1 H)- Found: 450.1275, tetrahydro-6-isoquinolinyl)-1 H- isoquinolinecarboxylate Rt: 2.52 min.
dihydro-2(1 H)- Found: 450.1275, tetrahydro-6-isoquinolinyl)-1 H- isoquinolinecarboxylate Rt: 2.52 min.
1 ,2,3-triazole-4-carboxamide MP: 275.7°C hydrochloride
(Intermediate 90)
Example 22: 1 -r(3.4-Dichlorophenyl)methyll-Λ/-(1.2.3.4-tetrahvdro-6-isoαuinolinyl)-1 H-1.2.3- triazole-4-carboxamide hydrochloride

HCI(g) was bubbled in EtOAc (1OmL) and a suspension of 1 ,1-dimethylethyl 6-[({1-[(3,4- dichlorophenyl)methyl]-1 H-1 ,2,3-triazol-4-yl}carbonyl)amino]-3,4-dihydro-2(1 H)- isoquinolinecarboxylate (Intermediate 91 ) (0.1 18g, 0.23 mmol) in EtOAc was added. The reaction mixture was stirred at room temperature overnight. The resulting precipitate was filtered and dried to give the title compound as a brown solid (68mg, 46%). HRMS (M+H)+: calculated for C19H17CI2N5O: 402.0888, Found: 402.0887, Rt: 2.41 min. MP: 294 0C
The following Example was similarly prepared by a method analogous to that described for Example 22:

Table 9

Example 25: N-{1-r(3.4-dichlorophenvnmethyll-5-methyl-1 H-1.2.3-triazol-4-yl)-1.2.3.4- tetrahydro-6-isoquinolinecarboxamide hydrochloride

This compound was similarly prepared by a method analogous to that described for Example 21 , starting from 1 ,1-dimethylethyl 6-[({1-[(3,4-dichlorophenyl)methyl]-5-methyl-1 H-1 ,2,3- triazol-4-yl}amino)carbonyl]-3,4-dihydro-2(1 H)-isoquinolinecarboxylate (Intermediate 94). HRMS (M+H)+: calculated for C20H19CI2N5O,Theo: 416,1045 found: 416.1021 , Rt: 2.46 min. MP: 156.7 0C
BIOLOGICAL ASSAY
The compounds of the present invention may be analysed in vitro for SCD activity using an assay based on the production of [3H]H2O, which is released during the enzyme-catalyzed generation of the monounsaturated fatty acyl CoA product. The assay is performed in a 96- well filtration plates. The titrated substrate used in the assay is the [9,10-3H] stearoyl Coenzyme A. After incubation for 6 minutes of SCD-containing rat microsomes (2 μg protein) and substrate (1 μM), the labelled fatty acid acyl-CoA species and microsomes are absorbed with charcoal and separated from [3H]H2O by centrifugation. The formation of [3H]H2O is used as a measure of SCD activity. Compounds at concentrations starting at 10 μM to 0.1 nM or vehicle (DMSO) are preincubated for 5 minutes with the microsomes before addition of the substrate. The concentration-responses are fitted with sigmoidal curves to obtain IC50 values.
All of the synthetic Example compounds tested by the above described in vitro assay for SCD activity were found to exhibit an average plC50 value equal to or greater than 5.5.
The following compound was prepared according to similar protocols to those described above for Example 1 and when tested by the above described in vitro assay for SCD activity was found to exhibit an average plC50 value in the range 5-5.5.
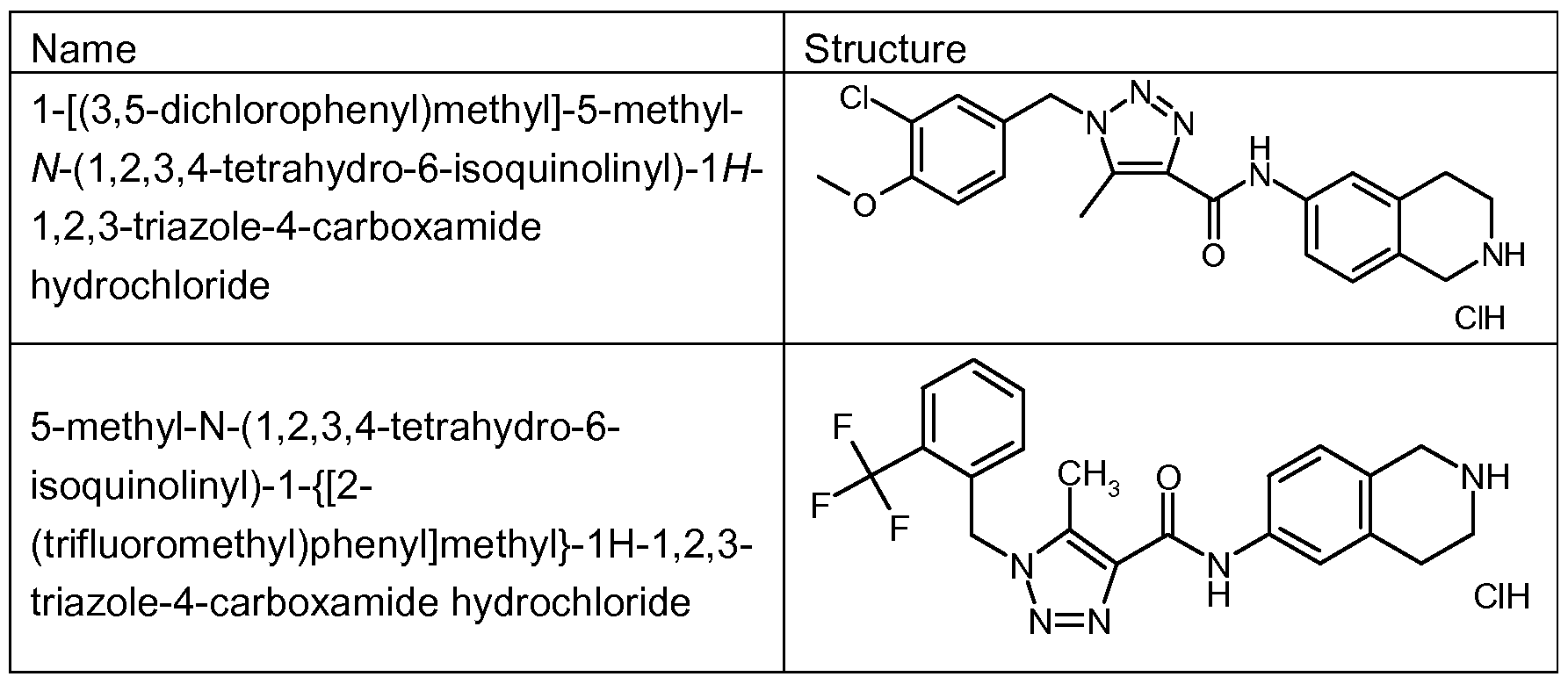
The following compounds were also prepared and when tested by the above described in vitro assay for SCD activity were found to exhibit an average plC50 value of less than 5.


Claims
1. A compound of formula (I):
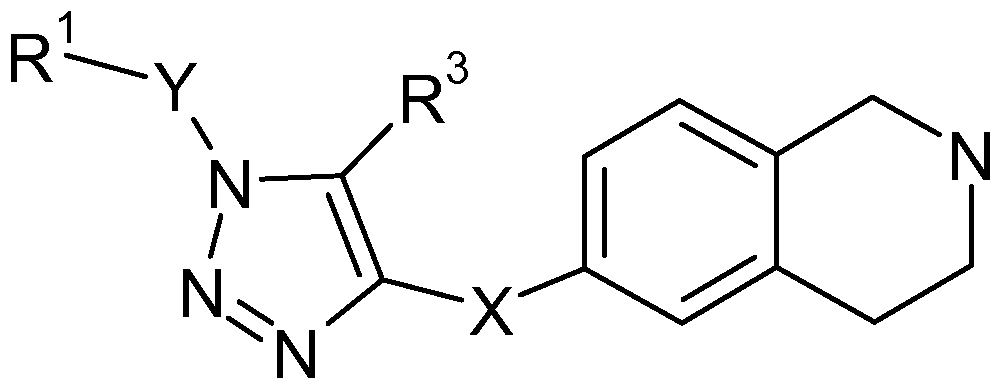
(I) wherein:
X represents -CONH- or -NHCO-,
R1 represents:
(i) a substituent selected from: -H or -Ci-6alkyl,
(N) -C6-ioaryl optionally substituted by one, two or three groups independently selected from:
(a) -C1-2alkyl, -C1-6haloalkyl or halogen,
(b) -C6-ioaryl, -C5-ioheteroaryl or -Cs-ioheterocyclyl, wherein the -C6-ioaryl, -C5- 10heteroaryl or -Cs-ioheterocyclyl ring is optionally substituted by one, two or three groups independently selected from: -C1-3alkyl, -C1-6alkoxy or -C1-6haloalkyl,
(iii) benzothiophene or thiophene wherein the benzothiophene or thiophene is optionally substituted by one, two or three groups independently selected from: -C1-6alkyl, -C1-6haloalkyl or halogen,
Y represents -CH2- or -OCH2-, R2 represents -H, and R3 represents -H or -Ci-2alkyl, or a salt thereof.
2. A compound of formula (I) or salt thereof according to claim 1 wherein X represents - CONH-.
3. A compound of formula (I) or salt thereof according to claim 1 or claim 2 wherein one of R1 represents -C6-ioaryl optionally substituted by: one, two or three groups independently selected from:
(a) -Ci-2alkyl, -Ci-6haloalkyl or halogen,
(b) -C6-ioaryl optionally substituted by one, two or three groups selected from: -C1-3alkyl or - C1-6haloalkyl.
4. A compound of formula (I) or salt thereof according to any one of claims 1 to 3 wherein Y represents -CH2-.
5. A compound of formula (I) or salt thereof according to any one of claims 1-4 wherein R3 represents H or -CH3.
6. A compound of formula (I) according to claim 1 selected from: 1-(2-Biphenylylmethyl)-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-4- carboxamide, 1-[(4-Fluorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1H-1 ,2,3-triazole-
4-carboxamide,
1-[(4-Bromophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 /-/-1 ,2,3-triazole-
4-carboxamide,
5-Methyl-1 -[(4-methylphenyl)methyl]-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole- 4-carboxamide,
1-[(2-Chlorophenyl)methyl]-5-methyl-Λ/-(1 !2!3!4-tetrahydro-6-isoquinolinyl)-1 H-1 !2!3-triazole-
4-carboxamide,
1-[(4-Chlorophenyl)methyl]-5-methyl-Λ/-(1 !2!3!4-tetrahydro-6-isoquinolinyl)-1 H-1 !2!3-triazole-
4-carboxamide, 1-[(3!4-Dichlorophenyl)methyl]-5-methyl-Λ/-(1 !2!3!4-tetrahydro-6-isoquinolinyl)-1 H-1 !2!3- triazole-4-carboxamide,
5-Methyl-1-(1-naphthalenylmethyl)-Λ/-(1 !2!3!4-tetrahydro-6-isoquinolinyl)-1 H-1 !2!3-triazole-4- carboxamide,
1 -[(3-Chlorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole- 4-carboxamide,
1-{[(2-Chlorophenyl)oxy]methyl}-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide,
5-Methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 -{[4-(trifluoromethyl)phenyl]methyl}-1 H-1 ,2,3- triazole-4-carboxamide, 1 -[(2,5-Dichlorophenyl)methyl]-5-methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide,
5-Methyl-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 -{[3-(trifluoromethyl)phenyl]methyl}-1 H-1 ,2,3- triazole-4-carboxamide, i-KS-Chloro-i-benzothien-S-yOmethyO-δ-methyl-N^I ^.S^-tetrahydro-θ-isoquinolinyO-I H- 1 ,2,3-triazole-4-carboxamide,
1-[(5-Chloro-2-thienyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide,
1-[(2,3-Dichlorophenyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide, 1-[(3,4-Difluorophenyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide,
1-(1-Benzothien-3-ylmethyl)-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-
4-carboxamide, i-P-Chloro-^fluorophenyOmethyll-S-methyl-N^I ^.S^-tetrahydro-θ-isoquinolinyO-I H-I ^.S- triazole-4-carboxamide,
1-[(3,5-Dichlorophenyl)methyl]-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide, 1-{[3-Chloro-4-(trifluoromethyl)phenyl]methyl}-5-methyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-
1 H-1 ,2,3-triazole-4-carboxamide,
1-[(3,4-Dichlorophenyl)methyl]-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3-triazole-4- carboxamide,
1-(1-Naphthalenylmethyl)-Λ/-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1H-1 ,2,3-triazole-4- carboxamide,
1-[(3,4-Dichlorophenyl)methyl]-5-ethyl-N-(1 ,2,3,4-tetrahydro-6-isoquinolinyl)-1 H-1 ,2,3- triazole-4-carboxamide, and
N-{1-[(3,4-Dichlorophenyl)methyl]-5-methyl-1 H-1 ,2,3-triazol-4-yl}-1 ,2,3,4-tetrahydro-6- isoquinoline carboxamide, or a salt thereof.
7. A compound of formula (I) or a salt thereof as claimed in any one of claims 1 to 6 wherein the salt is a pharmaceutically acceptable salt
8. A pharmaceutical composition comprising a compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6 together with at least one pharmaceutical carrier and/or excipient.
9. A compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6 for use in therapy.
10. Use of a compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6 for the manufacture of a medicament for treating and/or preventing a disease or a condition susceptible to amelioration by an SCD inhibitor.
11. Use of a compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6 for the manufacture of a medicament for treating and/or preventing diseases or conditions caused by or associated with an abnormal plasma lipid profile including dyslipidemia, hypoalphalipoproteinemia, hyperbetalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial hypercholesterolemia, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, atherosclerosis, obesity, Type I diabetes, Type Il diabetes, insulin resistance, hyperinsulinaemia and metabolic syndrome; peripheral vascular disease, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, thrombosis, hepatic steatosis, non-alcoholic steatohepatitis (NASH) and other diseases related to accumulation of lipids in the liver; eczema, acne, psoriasis, keloid scar formation or prevention, and diseases related to production or secretions from mucous membranes; cancer, neoplasia, malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like; mild cognitive impairment (MCI), Alzheimer's Disease (AD), cerebral amyloid angiopathy (CAA) or dementia associated with Down Syndrome (DS) and other neurodegenerative diseases characterized by the formation or accumulation of amyloid plaques comprising Aβ42.
12. Use of a compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6 for the manufacture of a medicament for treating and/or preventing acne, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or non-alcoholic steatohepatitis (NASH).
13. Use of a compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6 for the manufacture of a medicament for treating and/or preventing acne.
14. A compound of formula (I) or pharmaceutically acceptable salt according to any one of claims 1 to 6 for use in treating and/or preventing a disease or a condition susceptible to amelioration by an SCD inhibitor.
15. A compound of formula (I) or pharmaceutically acceptable salt according to any one of claims 1 to 6 for use in treating and/or preventing diseases or conditions caused by or associated with an abnormal plasma lipid profile including dyslipidemia, hypoalphalipoproteinemia, hyperbetalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial hypercholesterolemia, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, atherosclerosis, obesity, Type I diabetes, Type Il diabetes, insulin resistance, hyperinsulinaemia and metabolic syndrome; peripheral vascular disease, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, thrombosis, hepatic steatosis, non-alcoholic steatohepatitis (NASH) and other diseases related to accumulation of lipids in the liver; eczema, acne, psoriasis, keloid scar formation or prevention, and diseases related to production or secretions from mucous membranes; cancer, neoplasia, malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like; mild cognitive impairment (MCI), Alzheimer's Disease (AD), cerebral amyloid angiopathy (CAA) or dementia associated with Down Syndrome (DS) and other neurodegenerative diseases characterized by the formation or accumulation of amyloid plaques comprising Aβ42.
16. A compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6 for use in treating and/or preventing acne, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or non-alcoholic steatohepatitis (NASH).
17. A compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6 for use in treating and/or preventing acne.
18. A method of treating and/or preventing a disease or a condition susceptible to amelioration by an SCD comprising administering to a subject a therapeutically effective amount of a compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6.
19. A method of treating and/or preventing diseases or conditions caused by or associated with an abnormal plasma lipid profile including dyslipidemia, hypoalphalipoproteinemia, hyperbetalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial hypercholesterolemia, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, atherosclerosis, obesity, Type I diabetes, Type Il diabetes, insulin resistance, hyperinsulinaemia and metabolic syndrome; peripheral vascular disease, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, thrombosis, hepatic steatosis, non-alcoholic steatohepatitis (NASH) and other diseases related to accumulation of lipids in the liver; eczema, acne, psoriasis, keloid scar formation or prevention, and diseases related to production or secretions from mucous membranes; cancer, neoplasia, malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like; mild cognitive impairment (MCI), Alzheimer's Disease (AD), cerebral amyloid angiopathy (CAA) or dementia associated with Down Syndrome (DS) and other neurodegenerative diseases characterized by the formation or accumulation of amyloid plaques comprising Aβ42 comprising administering to a subject a therapeutically effective amount of compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6.
20 A method of treating and/or preventing acne, dyslipidemia, hypertriglyceridemia, atherosclerosis, obesity, Type Il diabetes, insulin resistance, hyperinsulinaemia, hepatic steatosis and/or non-alcoholic steatohepatitis (NASH) comprising administering to a subject a therapeutically effective amount of compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6.
21. A method of treating and/or preventing acne comprising administering to a subject a therapeutically effective amount of compound of formula (I) or pharmaceutically acceptable salt thereof according to any one of claims 1 to 6.
22. A compound of formula (I) or a pharmaceutically acceptable salt thereof in combination with one or more active agent(s) selected from an inhibitor of cholesteryl ester transferase (CETP inhibitors), a HMG-CoA reductase inhibitor, a microsomal triglyceride transfer protein, a peroxisome proliferator-activated receptor activator (PPAR), a bile acid reuptake inhibitor, a cholesterol absorption inhibitor, a cholesterol synthesis inhibitor, a fibrate, niacin, an ion-exchange resin, an antioxidant, an inhibitor of AcylCoA: cholesterol acyltransferase (ACAT inhibitor), a cannabinoid 1 antagonist, a bile acid sequestrant a corticosteroid, a vitamin D3 derivative, a retinoid, an immunomodulator, an anti androgen, a keratolytic agent, an anti-microbial, a platinum chemotherapeutic, an antimetabolite, hydroxyurea, a taxane, a mitotic disrupter, an anthracycline, dactinomycin, an alkylating agent and a cholinesterase inhibitor.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0715055.0A GB0715055D0 (en) | 2007-08-02 | 2007-08-02 | Compounds |
| GB0715055.0 | 2007-08-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009016216A1 true WO2009016216A1 (en) | 2009-02-05 |
Family
ID=38529168
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/060035 WO2009016216A1 (en) | 2007-08-02 | 2008-07-31 | Triazole derivatives as scd inhibitors |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB0715055D0 (en) |
| WO (1) | WO2009016216A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012030165A2 (en) | 2010-08-31 | 2012-03-08 | 서울대학교산학협력단 | Use of the fetal reprogramming of a ppar δ agonist |
| US8207204B2 (en) | 2007-11-09 | 2012-06-26 | Glaxosmithkline Llc | Triazole derivatives as SCD inhibitors |
| WO2013056148A2 (en) | 2011-10-15 | 2013-04-18 | Genentech, Inc. | Methods of using scd1 antagonists |
| US8486977B2 (en) | 2007-11-09 | 2013-07-16 | Glaxosmithkline Llc | 1,2,3-triazole derivatives for use as stearoyl-CoA desaturase inhibitors |
| US10973810B2 (en) | 2017-01-06 | 2021-04-13 | Yumanity Therapeutics, Inc. | Methods for the treatment of neurological disorders |
| US11873298B2 (en) | 2017-10-24 | 2024-01-16 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005011657A2 (en) * | 2003-07-30 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Piperazine derivatives and their use as therapeutic agents |
| WO2006101521A2 (en) * | 2004-09-20 | 2006-09-28 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
-
2007
- 2007-08-02 GB GBGB0715055.0A patent/GB0715055D0/en not_active Ceased
-
2008
- 2008-07-31 WO PCT/EP2008/060035 patent/WO2009016216A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005011657A2 (en) * | 2003-07-30 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Piperazine derivatives and their use as therapeutic agents |
| WO2006101521A2 (en) * | 2004-09-20 | 2006-09-28 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8207204B2 (en) | 2007-11-09 | 2012-06-26 | Glaxosmithkline Llc | Triazole derivatives as SCD inhibitors |
| US8486977B2 (en) | 2007-11-09 | 2013-07-16 | Glaxosmithkline Llc | 1,2,3-triazole derivatives for use as stearoyl-CoA desaturase inhibitors |
| US9051281B2 (en) | 2007-11-09 | 2015-06-09 | Glaxosmithkline Llc | Compounds |
| WO2012030165A2 (en) | 2010-08-31 | 2012-03-08 | 서울대학교산학협력단 | Use of the fetal reprogramming of a ppar δ agonist |
| WO2013056148A2 (en) | 2011-10-15 | 2013-04-18 | Genentech, Inc. | Methods of using scd1 antagonists |
| US9358250B2 (en) | 2011-10-15 | 2016-06-07 | Genentech, Inc. | Methods of using SCD1 antagonists |
| US10973810B2 (en) | 2017-01-06 | 2021-04-13 | Yumanity Therapeutics, Inc. | Methods for the treatment of neurological disorders |
| US11873298B2 (en) | 2017-10-24 | 2024-01-16 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| GB0715055D0 (en) | 2007-09-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2207770B1 (en) | 1, 2, 3-triazole derivatives for use as stearoyl-coa desaturase inhibitors | |
| EP2207774B1 (en) | Triazole derivatives as scd inhibitors | |
| EP2144895A2 (en) | Isoquinolinecarboxamides as inhibitors of stearoyl-coa desaturase (scd) | |
| EP2125799A1 (en) | Thiadiazole derivatives, inhibitors of stearoyl-coa desaturase | |
| WO2009056556A1 (en) | Substitute 1, 6-naphthyridines for use as scd inhibitors | |
| EP2099782A2 (en) | Isoquinolinecarboxamides as inhibitors of stearoyl-coa desaturase (scd) | |
| WO2009150196A1 (en) | N-thiazolyl-1, 2, 3, 4-tetrahydro-6-isoquinolinecarboxamide derivatives as inhibitors of stearoyl coenzyme a desaturase | |
| WO2009010560A1 (en) | Pyrazole derivatives and use thereof as inhibitors of stearoyl-coa desaturase | |
| EP2106398A2 (en) | Isoquinolinecarboxamides as inhibitors of stearoyl-coa desaturase (scd) | |
| WO2009016216A1 (en) | Triazole derivatives as scd inhibitors | |
| EP2102188A2 (en) | Compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08786660 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08786660 Country of ref document: EP Kind code of ref document: A1 |