WO2014116749A1 - Anti-hcv antibodies and methods of using thereof - Google Patents
Anti-hcv antibodies and methods of using thereof Download PDFInfo
- Publication number
- WO2014116749A1 WO2014116749A1 PCT/US2014/012610 US2014012610W WO2014116749A1 WO 2014116749 A1 WO2014116749 A1 WO 2014116749A1 US 2014012610 W US2014012610 W US 2014012610W WO 2014116749 A1 WO2014116749 A1 WO 2014116749A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antibody
- seq
- hcv
- amino acid
- acid sequence
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/42—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum viral
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/08—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses
- C07K16/081—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses from DNA viruses
- C07K16/082—Hepadnaviridae, e.g. hepatitis B virus
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Definitions
- the present invention relates to anti-HCV antibodies capable of inhibiting HCV infection and compositions comprising these antibodies, as well as methods for producing and using the same.
- HCV Hepatitis C virus
- HCC hepatocellular carcinoma
- New Direct Acting Antiviral (DAA) therapies such as viral protease and viral polymerase inhibitors, are currently under development for use as monotherapy or combination therapy and have the potential to replace the current SoC.
- DAA Direct Acting Antiviral
- effectiveness of such DAA therapies can be genotype specific and as a result such treatments are limited to subpopulations of HCV infected individuals.
- Six major HCV genotypes have been identified that are further divided into at least 70 different subtypes. Currently, HCV genotype 1 represents greater than 70% of infections in the US, Europe and Australia, and is the most difficult to cure.
- a significant challenge for the development of vaccines is the identification of protective epitopes that are conserved in the majority of viral genotypes and subtypes. This problem is compounded by the fact that the envelope proteins, the natural target for the neutralizing response, are two of the most variable proteins.
- HCV contains a positive-strand RNA genome which encodes a single polyprotein of approximately 3000 amino acids in length that is post-translationally processed to produce at least ten different proteins: core, envelope proteins El and E2, p7, and non- structural proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B.
- HCV entry into hepatocytes occurs through the coordinated interactions between the HCV E1-E2 heterodimer and at least four essential host cellular factors; CD81, scavenger receptor B type I (SR-BI), occludin (OCLN) and claudin 1 (CLDNl).
- SR-BI scavenger receptor B type I
- OCLN occludin
- CLDNl claudin 1
- the HCV E2 glycoprotein binds CD81 and SR-BI.
- the E2 glycoprotein extends from amino acids 384 to 746 of the polyprotein and has regions of extreme variability.
- an HCV-associated disorder or disease is HCV infection.
- an HCV-associated disorder or disease is liver disease.
- the invention provides an isolated antibody that binds hepatitis C virus E2 protein comprising a heavy chain variable region and a light chain variable region, (a) wherein the heavy chain variable region comprises three hypervariable region (HVR) sequences, wherein (i) HVR-Hl comprises GXiSX 2 TSGYWN (SEQ ID NO: l), wherein is D or E, and X2 is I or L; (ii) HVR-H2 comprises
- QQNNVDPWT (SEQ ID NO:6); wherein the antibody is not an antibody comprising (a) HVR-Hl comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 11 ; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 15; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising an amino acid sequence selected from SEQ ID NO:6.
- the antibody comprises HVR-Hl comprising the amino acid sequence of SEQ ID NO:7 or 8, HVR-H2 comprising the amino acid sequence of SEQ ID NO:9, 10 or 11, and HVR-H3 comprising the amino acid sequence of SEQ ID NO: 12 or 13.
- the antibody comprises HVR-L1 comprising the amino acid sequence of SEQ ID NO: 14, 15 or 16, HVR- L2 comprising the amino acid sequence of SEQ ID NO:5, and HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:7, HVR-H2 comprising the amino acid sequence of SEQ ID NO:9 or 10, and HVR-H3 comprising the amino acid sequence of SEQ ID NO: 12; and/or (b) HVR-Ll comprising the amino acid sequence of SEQ ID NO: 14, HVR-L2 comprising the amino acid sequence of SEQ ID NO:5, and HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the antibody comprises the heavy chain variable region sequence of SEQ ID NO: 17, 18, or 22, and/or the light chain variable region sequence of SEQ ID NO:24.
- the antibody comprises the heavy chain variable region sequence of SEQ ID NO: 19, 20, 22, or 23, and/or the light chain variable region sequence of SEQ ID NO:24 or 25. In some embodiments, the antibody comprises the heavy chain variable region sequence of SEQ ID NO:21, and/or the light chain variable region sequence of SEQ ID NO:24. In some embodiments, the antibody comprises the heavy chain sequence of SEQ ID NO:27 or 28, and the light chain sequence of SEQ ID NO:30. In some embodiments herein, the antibody is a humanized antibody. In some embodiments herein, the antibody is an antigen-binding fragment. In some
- the antibody is an antibody fragment selected from the group consisting of a Fab, Fab'-SH, Fv, scFv, and (Fab') 2 fragment.
- the antibody inhibits infection of one or more HCV genotypes (such as genotype la, genotype lb, genotype 2a, and genotype 4a) to liver cells.
- the invention provides an isolated antibody that binds hepatitis C virus E2 protein comprising a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising amino acid sequence SGYWN (SEQ ID NO:33), CDR H2 comprising the amino acid sequence of SEQ ID NO:9, 10, or 11, CDR H3 comprising amino acid sequence ITTSTYAMDY (SEQ ID NO:34) or ITTTTYAMDY (SEQ ID NO:35); and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, 15, or 16, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- the heavy chain variable region comprises CDR HI comprising amino acid sequence SGYWN (SEQ ID NO:33), CDR H2 comprising the amino acid sequence of SEQ ID NO:9, 10, or 11, CDR H3 comprising amino acid sequence ITTSTYAMDY (SEQ ID
- the anti-HCV antibody is not an antibody comprising a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR
- the antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO:9 or 10, CDR H3 comprising the amino acid sequence of SEQ ID NO:34; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- the antibody is a humanized antibody.
- the antibody is an antigen-binding fragment.
- the antibody is an antibody fragment selected from the group consisting of a Fab, Fab'-SH, Fv, scFv, and (Fab') 2 fragment.
- the antibody inhibits infection of one or more HCV genotypes (such as genotype la, genotype lb, genotype 2a, and genotype 4a) to liver cells.
- the invention provides an antibody that binds hepatitis C virus E2 protein produced by a method comprising culturing a host cell comprising a nucleic acid encoding any of the antibodies disclosed herein under a condition suitable for expression of the nucleic acid; and recovering the antibody produced by the cell.
- the host cell comprises a nucleic acid encoding a VL amino acid sequence, and/or a nucleic acid encoding a VH amino acid sequence of any of the anti-HCV antibodies described above and herein.
- the invention provides a pharmaceutical composition comprising any of the antibodies disclosed herein, and a pharmaceutically acceptable carrier.
- the invention provides a kit or an article of manufacture comprising one or more of the anti-HCV antibodies described above and herein.
- the kit or article of manufacture may further comprise a label or package insert indicating that the antibody is used for the treatment, prevention and/or diagnosis of the disorders described herein (e.g., hepatitis C virus infection).
- the kit or article of manufacture may further comprise a second therapeutic agent.
- the invention also provides an isolated nucleic acid encoding any of the antibodies disclosed herein.
- the invention provides a vector comprising a nucleic acid encoding any of the antibodies disclosed herein.
- the vector is an expression vector.
- the vector comprises a nucleic acid encoding a VL amino acid sequence of any of the anti-HCV antibodies described above and herein.
- the vector comprises a nucleic acid encoding a VH amino acid sequence of any of the anti-HCV antibodies described above and herein.
- the invention also provides a host cell comprising a nucleic acid encoding any of the antibodies disclosed herein.
- the host cell is prokaryotic or eukaryotic.
- the host cell comprises a nucleic acid encoding a VL amino acid sequence, and/or a nucleic acid encoding a VH amino acid sequence of any of the anti-HCV antibodies described above and herein.
- the invention provides a method of producing an antibody comprising culturing a host cell comprising a nucleic acid encoding any of the antibodies disclosed herein under a condition suitable for expression of the nucleic acid; and recovering the antibody produced by the cell.
- the method further comprises purifying the antibody.
- the host cell comprises a nucleic acid encoding a VL amino acid sequence, and/or a nucleic acid encoding a VH amino acid sequence of any of the anti-HCV antibodies described above and herein.
- the invention provides a method for treating or preventing a hepatitis C virus infection in a subject, comprising administering to the subject an effective amount of any of the antibodies disclosed herein.
- the subject is a human.
- the subject has been diagnosed with the hepatitis C virus infection.
- the hepatitis C virus infection is an acute hepatitis C virus infection.
- the hepatitis C virus infection is a chronic hepatitis C virus infection.
- treating the hepatitis C virus infection comprises reducing viral load.
- the method further comprises administering a second therapeutic agent.
- the second therapeutic agent is a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a cyclophilin inhibitor, an antibody that targets a non-E2 HCV protein, or a combination thereof.
- the second therapeutic agent is a HCV protease inhibitor selected from the group consisting of Teleprevir, Boceprevir, Semeprevir (TMC435), BI201335, Danoprevir/r (RG7227), Vaniprevir (MK-7009), Narlaprevir (SCH 900518), Asunaprevir, GS-9256, GS-9451, ABT-450/r, ACH-1625, ACH-2684, and MK-5172.
- the second therapeutic agent is a polymerase inhibitor selected from the group consisting of PSI-7977, Mercitabine, IDX184, PSI-938, INX-189, Tegobuvir, Filibuvir, Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055.
- a polymerase inhibitor selected from the group consisting of PSI-7977, Mercitabine, IDX184, PSI-938, INX-189, Tegobuvir, Filibuvir, Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055.
- the treatment is an interferon-free treatment.
- the subject has sustained virologic response for at least 12 weeks after stopping the treatment.
- the viral load in the subject has been reduced to an undetectable level after the treatment.
- the viral resistance in the subject is undetectable or low.
- the subject is not responsive to an interferon treatment.
- the invention provides a method for treating or preventing a hepatitis C virus infection in a subject, comprising administering to the subject an effective amount of an antibody that specifically binds hepatitis C virus E2 protein and a second therapeutic agent.
- the invention provides a method of preventing developing resistance to treatment, comprising administering an effective amount of an antibody that specifically binds hepatitis C virus E2 protein and a second therapeutic agent.
- the second therapeutic agent is a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a cyclophilin inhibitor, an antibody that targets a non-E2 HCV protein, and a combination thereof.
- the antibody comprises a heavy chain variable region and a light chain variable region, (a) wherein the heavy chain variable region comprises three hypervariable region (HVR) sequences, wherein (i) HVR-H1 comprises GXiSX 2 TSGYWN (SEQ ID NO: l), wherein ⁇ is D or E, and X2 is I or L; (ii) HVR-H2 comprises YISYSGSTYYXiX 2 SLRS (SEQ ID NO:2), wherein X 1 is N or S, and X 2 is P or L; and (iii) HVR-H3 comprises
- HVR-L1 comprises
- HVR- L2 comprises LASNLNS (SEQ ID NO:5)
- HVR-L3 comprises QQNNVDPWT (SEQ ID NO:6).
- the invention provides a method of preventing of HCV infection of a transplanted liver, comprising administering to the subject an effective amount of any of the antibodies disclosed herein before, during or after the subject receives the liver transplant.
- the invention also provides use of an anti-HCV antibody described above and herein in the manufacture of a medicament for use in any of the methods described above and herein (e.g., for treating or preventing a hepatitis C virus infection in a subject, for preventing developing resistance to treatment, or preventing HCV infection of a transplanted liver).
- the invention also provides an anti-HCV antibody described above and herein for use in any of the methods described above and herein (e.g., for treating or preventing a hepatitis C virus infection in a subject, for preventing developing resistance to treatment, or preventing HCV infection of a transplanted liver).
- Figure 1 is a diagram of the HCV E2 protein epitope binding site.
- Figure 2 shows an amino acid sequence alignment of the A) light chain variable region and B) heavy chain variable region of MRCT10.1, V361, and V362 numbered according to Kabat. Boxed amino acid sequences indicate hypervariable regions (HVRs). Bars over the sequence indicate the complementarity determining regions (CDRs). From the N-terminus to C-terminus (left to right), amino acid sequences outside of boxed HVRs indicate FR1, FR2, FR3, and FR4.
- “MRCT10" in the figure refers to the MRCT10.1 amino acid sequence.
- FIG. 3 is a series of graphs showing binding of V362 to wild-type (WT) and
- FIG 4 is a graph depicting enhancement of the antiviral effect of an NS3 protease inhibitor, Telaprevir, with an anti-HCV antibody.
- HCV RNA copies were measured at the indicated times post infection.
- HCV cDNA derived from the day 22 post-infection cultures was sequenced. Dotted line represents the limit of qPCR linear range. Representative data from two independent experiments.
- Figure 5 is a series of graphs depicting enhancement of the antiviral effect of Telaprevir and interferon combination treatment with anti-HCV antibodies.
- A) DMSO- differentiated Huh-7.5 cells were infected with Jcl HCVcc (MOI 0.05) alone (filled squares) or in the presence 0.3 ⁇ Telaprevir plus 5 IU/ml Interferon alpha (IFN-a) (open circles) or a combination of 0.3 ⁇ Telaprevir and 5 IU/ml IFN-a plus V335 (10 ⁇ g/mL, filled inverted diamonds).
- Asterisks denote undetectable HCV cDNA in cultures treated with a combination of V335, Telaprevir, and IFN-a at 10, 14, 22, and 30 days post infection. Representative data from two independent experiments. Dotted line represents the limit of qPCR linear range.
- B) DMSO-differentiated Huh-7.5 cells were infected with Jcl HCVcc (MOI 0.05) alone (filled squares) or in the presence 0.3 ⁇ Telaprevir plus 5 IU/ml Interferon alpha (IFN-a) (open circles) or a combination of 0.3 ⁇ Telaprevir and 5 IU/ml IFN-a plus MRCT10.1 (10 ⁇ g/mL, filled inverted diamonds). HCV RNA copies were measured at various times post infection by RT-qPCR. Representative data from two independent experiments. Dotted line represents the limit of qPCR linear range.
- Anti-HCV antibodies that bind HCV E2 protein
- Anti-HCV antibodies of the present invention can be used as a therapeutic agent for use in the treatment of HCV-associated diseases.
- an "acceptor human framework” for the purposes herein is a framework comprising the amino acid sequence of a light chain variable domain (VL) framework or a heavy chain variable domain (VH) framework derived from a human immunoglobulin framework or a human consensus framework, as defined below.
- An acceptor human framework "derived from” a human immunoglobulin framework or a human consensus framework may comprise the same amino acid sequence thereof, or it may contain amino acid sequence changes. In some embodiments, the number of amino acid changes are 10 or less, 9 or less, 8 or less, 7 or less, 6 or less, 5 or less, 4 or less, 3 or less, or 2 or less.
- the VL acceptor human framework is identical in sequence to the VL human immunoglobulin framework sequence or human consensus framework sequence.
- binding affinity refers to the strength of the sum total of noncovalent interactions between a single binding site of a molecule (e.g., an antibody) and its binding partner (e.g., an antigen).
- binding affinity refers to intrinsic binding affinity which reflects a 1: 1 interaction between members of a binding pair (e.g., antibody and antigen).
- the affinity of a molecule X for its partner Y can generally be represented by the dissociation constant (Kd). Affinity can be measured by common methods known in the art, including those described herein. Specific illustrative and exemplary embodiments for measuring binding affinity are described in the following.
- An "affinity matured” antibody refers to an antibody with one or more alterations in one or more hypervariable regions (HVRs), compared to a parent antibody which does not possess such alterations, such alterations resulting in an improvement in the affinity of the antibody for antigen.
- HVRs hypervariable regions
- anti-HCV antibody an antibody that binds to HCV
- an antibody that binds to HCV E2 protein refers to an antibody that is capable of binding to HCV E2 protein with sufficient affinity such that the antibody is useful as a diagnostic and/or therapeutic agent in targeting HCV E2 protein and/or HCV.
- the extent of binding of an anti-HCV antibody to an unrelated, non-HCV E2 protein is less than about 10% of the binding of the antibody to HCV E2 protein and/or HCV as measured, e.g., by a radioimmunoassay (RIA).
- RIA radioimmunoassay
- an antibody that binds to HCV E2 protein and/or HCV has a dissociation constant (Kd) of ⁇ ⁇ , ⁇ 100 nM, ⁇ 10 nM, ⁇ 1 nM, ⁇ 0.1 nM, ⁇ 0.01 nM, or ⁇ 0.001 nM (e.g. 10 "8 M or less, e.g. from 10 "8 M to 10 "13 M, e.g., from 10 "
- an anti-HCV antibody binds to an epitope of HCV E2 protein that is conserved among HCV from different genotypes.
- antibody herein is used in the broadest sense and encompasses various antibody structures, including but not limited to monoclonal antibodies, polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired antigen-binding activity.
- an "antibody fragment” refers to a molecule other than an intact antibody that comprises a portion of an intact antibody that binds the antigen to which the intact antibody binds.
- antibody fragments include but are not limited to Fv, Fab, Fab', Fab'-SH, F(ab') 2 ; diabodies; linear antibodies; single-chain antibody molecules (e.g. scFv); and multispecific antibodies formed from antibody fragments.
- an "antibody that binds to the same epitope" as a reference antibody refers to an antibody that blocks binding of the reference antibody to its antigen in a competition assay by 50% or more, and conversely, the reference antibody blocks binding of the antibody to its antigen in a competition assay by 50% or more.
- chimeric antibody refers to an antibody in which a portion of the heavy and/or light chain is derived from a particular source or species, while the remainder of the heavy and/or light chain is derived from a different source or species.
- the "class" of an antibody refers to the type of constant domain or constant region possessed by its heavy chain.
- the heavy chain constant domains that correspond to the different classes of immunoglobulins are called ⁇ , ⁇ , ⁇ , ⁇ , and ⁇ , respectively.
- cytotoxic agent refers to a substance that inhibits or prevents a cellular function and/or causes cell death or destruction. Cytotoxic agents include, but are not limited to, radioactive isotopes (e.g., At 211 , 1 131 , 1 125 , Y 90 , Re 186 , Re 188 , Sm 153 ,
- Bi , P , Pb and radioactive isotopes of Lu chemotherapeutic agents or drugs (e.g., methotrexate, adriamicin, vinca alkaloids (vincristine, vinblastine, etoposide), doxorubicin, melphalan, mitomycin C, chlorambucil, daunorubicin or other intercalating agents); growth inhibitory agents; enzymes and fragments thereof such as nucleolytic enzymes; antibiotics; toxins such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, including fragments and/or variants thereof; and the various antitumor or anticancer agents disclosed below.
- chemotherapeutic agents or drugs e.g., methotrexate, adriamicin, vinca alkaloids (vincristine, vinblastine, etoposide), doxorubicin, melphalan, mitomycin C, chloramb
- Antibody effector functions refer to those biological activities attributable to the Fc region of an antibody, which vary with the antibody isotype. Examples of antibody effector functions include: Clq binding and complement dependent cytotoxicity (CDC); Fc receptor binding; antibody-dependent cell-mediated cytotoxicity (ADCC); phagocytosis; down regulation of cell surface receptors (e.g. B cell receptor); and B cell activation.
- an "effective amount" of an agent refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired therapeutic or prophylactic result.
- An effective amount can be provided in one or more administrations.
- a “therapeutically effective amount” is at least the minimum concentration required to effect a measurable improvement of a particular disorder (e.g., HCV infection).
- a therapeutically effective amount herein may vary according to factors such as the disease state, age, sex, and weight of the patient, and the ability of the anti-HCV antibody to elicit a desired response in the individual.
- a therapeutically effective amount is also one in which any toxic or detrimental effects of the anti-HCV antibody are outweighed by the
- prophylactically effective amount refers to an amount effective, at the dosages and for periods of time necessary, to achieve the desired prophylactic result. Typically but not necessarily, since a prophylactic dose is used in subjects prior to or at an earlier stage of disease, a prophylactically effective amount may be less than a therapeutically effective amount.
- Fc region herein is used to define a C-terminal region of an immunoglobulin heavy chain, including native- sequence Fc regions and variant Fc regions.
- the human IgG heavy-chain Fc region is usually defined to stretch from an amino acid residue at position Cys226, or from Pro230, to the carboxyl-terminus thereof.
- the C-terminal lysine (residue 447 according to the EU numbering system) of the Fc region may be removed, for example, during production or purification of the antibody, or by recombinantly engineering the nucleic acid encoding a heavy chain of the antibody. Accordingly, a composition of intact antibodies may comprise antibody populations with all K447 residues removed, antibody populations with no K447 residues removed, and antibody populations having a mixture of antibodies with and without the K447 residue.
- Suitable native- sequence Fc regions for use in the antibodies of the invention include human IgGl, IgG2, IgG3 and IgG4. Unless otherwise specified herein, numbering of amino acid residues in the Fc region or constant region is according to the EU numbering system, also called the EU index, as described in Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD, 1991.
- FR refers to variable domain residues other than hypervariable region (HVR) residues.
- the FR of a variable domain generally consists of four FR domains: FR1, FR2, FR3, and FR4. Accordingly, the HVR and FR sequences generally appear in the following sequence in VH (or VL): FR1-H1(L1)-FR2-H2(L2)-FR3-H3(L3)-FR4.
- full length antibody “intact antibody,” and “whole antibody” are used herein interchangeably to refer to an antibody having a structure substantially similar to a native antibody structure or having heavy chains that contain an Fc region as defined herein.
- Host cells include “transformants” and “transformed cells,” which include the primary transformed cell and progeny derived therefrom without regard to the number of passages.
- Progeny may not be completely identical in nucleic acid content to a parent cell, but may contain mutations. Mutant progeny that have the same function or biological activity as screened or selected for in the originally transformed cell are included herein.
- a "human antibody” is one which possesses an amino acid sequence which corresponds to that of an antibody produced by a human or a human cell or derived from a non-human source that utilizes human antibody repertoires or other human antibody- encoding sequences. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen-binding residues.
- a "human consensus framework” is a framework which represents the most commonly occurring amino acid residues in a selection of human immunoglobulin VL or VH framework sequences.
- the selection of human immunoglobulin VL or VH sequences is from a subgroup of variable domain sequences.
- the subgroup of sequences is a subgroup as in Kabat et al., Sequences of Proteins of Immunological Interest, Fifth Edition, NIH Publication 91-3242, Bethesda MD (1991), vols. 1-3.
- the subgroup is subgroup kappa I as in Kabat et al., supra.
- the subgroup is subgroup III as in Kabat et al., supra.
- a “humanized” antibody refers to a chimeric antibody comprising amino acid residues from non-human HVRs and amino acid residues from human FRs.
- a humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the HVRs (e.g., CDRs) correspond to those of a non-human antibody, and all or substantially all of the FRs correspond to those of a human antibody.
- a humanized antibody optionally may comprise at least a portion of an antibody constant region derived from a human antibody.
- a "humanized form" of an antibody, e.g., a non-human antibody refers to an antibody that has undergone humanization.
- hypervariable region refers to each of the regions of an antibody variable domain which are hypervariable in sequence and/or form structurally defined loops ("hypervariable loops").
- native four-chain antibodies comprise six HVRs; three in the VH (HI, H2, H3), and three in the VL (LI, L2, L3).
- HVRs generally comprise amino acid residues from the hypervariable loops and/or from the
- CDRs complementarity determining regions
- An HVR region as used herein comprise any number of residues located within positions 24-36 (for LI), 46-56 (for L2), 89-97 (for L3), 26-35B (for HI), 47-65 (for H2), and 93-102 (for H3). Therefore, an HVR includes residues in positions described previously:
- CDRs generally comprise the amino acid residues that form the hypervariable loops.
- CDRs also comprise "specificity determining residues,” or "SDRs,” which are residues that contact antigen. SDRs are contained within regions of the CDRs called abbreviated-CDRs, or a-CDRs.
- Exemplary a-CDRs (a-CDR-Ll, a-CDR-L2, a-CDR-L3, a-CDR-Hl, a-CDR-H2, and a-CDR-H3) occur at amino acid residues 31-34 of LI, 50-55 of L2, 89-96 of L3, 31-35B of HI, 50-58 of H2, and 95-102 of H3.
- HVR residues and other residues in the variable domain are numbered herein according to Kabat et al., supra.
- an “immunoconjugate” is an antibody conjugated to one or more heterologous molecule(s), including but not limited to a cytotoxic agent.
- mammals include, but are not limited to, domesticated animals (e.g., cows, sheep, cats, dogs, and horses), primates (e.g., humans and non-human primates such as monkeys), rabbits, and rodents (e.g., mice and rats).
- domesticated animals e.g., cows, sheep, cats, dogs, and horses
- primates e.g., humans and non-human primates such as monkeys
- rabbits e.g., mice and rats
- rodents e.g., mice and rats.
- the individual or subject is a human.
- an "isolated" antibody is one which has been separated from a component of its natural environment.
- an antibody is purified to greater than 95% or 99% purity as determined by, for example, electrophoretic (e.g., SDS-PAGE, isoelectric focusing (IEF), capillary electrophoresis) or chromatographic (e.g., ion exchange or reverse phase HPLC).
- electrophoretic e.g., SDS-PAGE, isoelectric focusing (IEF), capillary electrophoresis
- chromatographic e.g., ion exchange or reverse phase HPLC
- An "isolated" nucleic acid refers to a nucleic acid molecule that has been separated from a component of its natural environment.
- An isolated nucleic acid includes a nucleic acid molecule contained in cells that ordinarily contain the nucleic acid molecule, but the nucleic acid molecule is present extrachromosomally or at a chromosomal location that is different from its natural chromosomal location.
- isolated nucleic acid encoding an anti-HCV antibody refers to one or more nucleic acid molecules encoding antibody heavy and light chains (or fragments thereof), including such nucleic acid molecule(s) in a single vector or separate vectors, and such nucleic acid molecule(s) present at one or more locations in a host cell.
- the term "monoclonal antibody” as used herein refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical and/or bind the same epitope, except for possible variant antibodies, e.g., containing naturally occurring mutations or arising during production of a monoclonal antibody preparation, such variants generally being present in minor amounts.
- polyclonal antibody preparations typically include different antibodies directed against different determinants (epitopes)
- each monoclonal antibody of a monoclonal antibody preparation is directed against a single determinant on an antigen.
- the modifier "monoclonal” indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method.
- the monoclonal antibodies to be used in accordance with the present invention may be made by a variety of techniques, including but not limited to the hybridoma method, recombinant DNA methods, phage-display methods, and methods utilizing transgenic animals containing all or part of the human immunoglobulin loci, such methods and other exemplary methods for making monoclonal antibodies being described herein.
- a “naked antibody” refers to an antibody that is not conjugated to a heterologous moiety (e.g., a cytotoxic moiety) or radiolabel.
- the naked antibody may be present in a pharmaceutical formulation.
- Native antibodies refer to naturally occurring immunoglobulin molecules with varying structures.
- native IgG antibodies are heterotetrameric glycoproteins of about 150,000 daltons, composed of two identical light chains and two identical heavy chains that are disulfide-bonded. From N- to C-terminus, each heavy chain has a variable region (VH), also called a variable heavy domain or a heavy chain variable domain, followed by three constant domains (CHI, CH2, and CH3).
- VH variable region
- VL variable region
- the light chain of an antibody may be assigned to one of two types, called kappa ( ⁇ ) and lambda ( ⁇ ), based on the amino acid sequence of its constant domain.
- package insert is used to refer to instructions customarily included in commercial packages of therapeutic products, that contain information about the indications, usage, dosage, administration, combination therapy, contraindications and/or warnings concerning the use of such therapeutic products.
- Percent ( ) amino acid sequence identity with respect to a reference polypeptide sequence is defined as the percentage of amino acid residues in a candidate sequence that are identical with the amino acid residues in the reference polypeptide sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity. Alignment for purposes of determining percent amino acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or Megalign (DNASTAR) software. Those skilled in the art can determine appropriate parameters for aligning sequences, including any algorithms needed to achieve maximal alignment over the full length of the sequences being compared.
- % amino acid sequence identity values are generated using the sequence comparison computer program ALIGN-2.
- the ALIGN-2 sequence comparison computer program was authored by Genentech, Inc., and the source code has been filed with user documentation in the U.S. Copyright Office,
- the ALIGN-2 program is publicly available from Genentech, Inc., South San Francisco, California, or may be compiled from the source code.
- the ALIGN-2 program should be compiled for use on a UNIX operating system, including digital UNIX V4.0D. All sequence comparison parameters are set by the ALIGN-2 program and do not vary.
- % amino acid sequence identity of a given amino acid sequence A to, with, or against a given amino acid sequence B is calculated as follows:
- pharmaceutical formulation refers to a preparation which is in such form as to permit the biological activity of an active ingredient contained therein to be effective, and which contains no additional components which are unacceptably toxic to a subject to which the formulation would be administered.
- pharmaceutically acceptable carrier refers to an ingredient in a pharmaceutical formulation, other than an active ingredient, which is nontoxic to a subject.
- pharmaceutically acceptable carrier includes, but is not limited to, a buffer, excipient, stabilizer, or preservative.
- HCV E2 protein refers to any native HCV E2 protein isolated from or identified in any vertebrate source, including mammals such as primates (e.g. humans and chimpanzees) and rodents (e.g., mice and rats) unless otherwise indicated.
- the term encompasses "full-length,” unprocessed HCV E2 protein as well as any form of HCV E2 protein that results from processing in the cell or processing outside of the cell.
- the term also encompasses naturally occurring variants of HCV E2 protein, e.g., genotype variants or quasispecies. Exemplary naturally occurring variants of HCV E2 proteins can be found in, but not limited to, Simmonds P., J Gen Virol., (2004)., 85 (Pt 11):3173-88.
- treatment refers to clinical intervention designed to alter the natural course of the individual or cell being treated during the course of clinical pathology. Desirable effects of treatment include, but are not limited to, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, decreasing the rate of disease
- antibodies of the invention are used to delay development of a disease or to slow the progression of a disease.
- the disease is an HCV-associated disease.
- the HCV-associated disease is HCV infection. An individual is successfully "treated", for example, if one or more symptoms associated with HCV infection are mitigated or eliminated.
- prevention includes providing prophylaxis with respect to occurrence or recurrence of a disease in an individual.
- An individual may be predisposed to, susceptible to an HCV-associated disorder, or at risk of developing an HCV-associated disorder, but has not yet been diagnosed with the disorder.
- an HCV- associated disorder is HCV infection.
- an HCV-associated disorder is hepatocellular carcinoma.
- variable region refers to the domain of an antibody heavy or light chain that is involved in binding the antibody to antigen.
- the variable domains of the heavy chain and light chain (VH and VL, respectively) of a native antibody generally have similar structures, with each domain comprising four conserved framework regions (FRs) and three hypervariable regions (HVRs).
- FRs conserved framework regions
- HVRs hypervariable regions
- antibodies that bind a particular antigen may be isolated using a VH or VL domain from an antibody that binds the antigen to screen a library of complementary VL or VH domains, respectively. See, e.g., Portolano et al., J. Immunol. 150:880-887 (1993); Clarkson et al., Nature 352:624-628 (1991).
- vector refers to a nucleic acid molecule capable of propagating another nucleic acid to which it is linked.
- the term includes the vector as a self- replicating nucleic acid structure as well as the vector incorporated into the genome of a host cell into which it has been introduced.
- Certain vectors are capable of directing the expression of nucleic acids to which they are operatively linked. Such vectors are referred to herein as "expression vectors.”
- the invention provides methods for inhibiting, treating or preventing hepatitis C virus (HCV) infection in an individual comprising administering to the individual an effective amount of an anti-HCV antibody described herein.
- an effective amount of an anti-HCV antibody is administered to an individual for inhibiting, treating or preventing HCV cellular entry in an individual.
- an effective amount of an anti-HCV antibody is administered to an individual for inhibiting, treating or preventing HCV spread in an individual.
- an effective amount of an anti-HCV antibody is administered to an individual for inhibiting, treating or preventing an HCV-associated disease in the individual.
- the HCV is a drug-resistant HCV.
- compositions comprising one or more of those agents.
- Such compositions may further comprise suitable excipients, such as pharmaceutically acceptable excipients (carriers) including buffers, acids, bases, sugars, diluents, preservatives, and the like, which are well known in the art and are described herein.
- carriers such as buffers, acids, bases, sugars, diluents, preservatives, and the like, which are well known in the art and are described herein.
- carriers including buffers, acids, bases, sugars, diluents, preservatives, and the like, which are well known in the art and are described herein.
- carriers including buffers, acids, bases, sugars, diluents, preservatives, and the like, which are well known in the art and are described herein.
- present methods can be used alone or in combination with other conventional methods of treatment (e.g., antivirals).
- the methods of the present invention use an anti-HCV antibody, which term refers to an anti-HCV antibody that binds to HCV E2 protein.
- the HCV E2 protein is expressed on a hepatitis C viral surface and therefore inhibits HCV cellular entry by preventing binding of HCV E2 protein to a receptor expressed on a host cell surface (such as CD81 expressed on a human cell surface). See Figure 1.
- the anti-HCV antibodies described herein may have one or more of the following characteristics: (a) bind HCV E2 protein or variants thereof (such as HCV E2 glycosylation variants); (b) block binding of HCV E2 protein to a host cell (such as a human host cell); (c) inhibit HCV entry into a host cell; (d) inhibit and/ or prevent HCV infection of a host cell (such as a host cell in an individual); (e) inhibit HCV spread in an individual (such as a human); (f) treat and/or prevent emergence of drug-resistant HCV; (g) enhance inhibition of HCV infection and/or spread by other antivirals (such as HCV protease inhibitors); and (h) treat and/or prevent an HCV-associated disease (such as hepatocellular carcinoma).
- the activities of anti-HCV antibodies may be measured in vitro and/or in vivo.
- the invention provides an anti-HCV antibody comprising at least one, two, three, four, five, or six hypervariable regions (HVRs) selected from (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: l; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:2; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO:3; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO:4; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- HVRs hypervariable regions
- the invention provides an anti-HCV antibody comprising at least one, at least two, or all three VH HVR sequences selected from (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: l; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:2; and (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO:3.
- the anti-HCV antibody comprises HVR-H3 comprising the amino acid sequence of SEQ ID NO:3.
- the anti-HCV antibody comprises HVR- H3 comprising the amino acid sequence of SEQ ID NO:3 and HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the anti-HCV antibody comprises HVR-H3 comprising the amino acid sequence of SEQ ID NO:3, HVR-L3 comprising the amino acid sequence of SEQ ID NO:6, and HVR-H2 comprising the amino acid sequence of SEQ ID NO:2.
- the anti-HCV antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: l ; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:2; and (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO:3.
- the invention provides an anti-HCV antibody comprising at least one, at least two, or all three VH HVR sequences selected from (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:4; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (c) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the anti-HCV antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:4; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (c) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the anti-HCV antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: l ; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:2; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO:3; (d) HVR- Ll comprising the amino acid sequence of SEQ ID NO:4; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising an amino acid sequence selected from SEQ ID NO:6.
- SEQ ID NO: 1 refers to amino acid sequence
- SEQ ID NO:2 refers to amino acid sequence YISYSGSTYYXiX 2 SLRS, wherein ⁇ is N or S, and wherein X 2 is P or L.
- SEQ ID NO:3 refers to amino acid sequence ALITTXiTYAMDY, wherein X l is S or T.
- SEQ ID NO:4 refers to amino acid sequence RASES VX 1 GYGX 2 SFLH, wherein X l is D or S, and X 2 is N or Y.
- SEQ ID NO:5 refers to amino acid sequence LASNLNS.
- SEQ ID NO:6 refers to amino acid sequence QQNNVDPWT.
- the anti-HCV antibody is not an antibody comprising (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 11 ; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 15; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising an amino acid sequence selected from SEQ ID NO:6.
- an anti-HCV antibody of the invention comprises a heavy chain variable region and a light chain variable region,
- HVR-Hl comprising GXiSX 2 TSGYWN (SEQ ID NO: l), wherein Xi is D or E, and X 2 is I or L;
- HVR-H2 comprising YISYSGSTYYXiX 2 SLRS (SEQ ID NO:2), wherein X l is N or S, and wherein X 2 is P or L;
- HVR-H3 comprising ALITTXiTYAMDY (SEQ ID NO:3), wherein Xi is S or T; and/or
- HVR-Ll comprising RASES VXiGYGX 2 SFLH (SEQ ID NO:4), wherein Xi is D or S, and X 2 is N or Y;
- HVR-L2 comprising LASNLNS (SEQ ID NO:5);
- HVR-L3 comprising QQNNVDPWT (SEQ ID NO:6).
- an anti-HCV antibody of the invention comprises a heavy chain variable region and a light chain variable region
- HVR-Hl comprising GDSITSGYWN (SEQ ID NO:7) or GESLTSGYWN (SEQ ID NO:8);
- HVR-H2 comprising YISYSGSTYYNPSLRS (SEQ ID NO:9),
- HVR-H3 comprising ALITTSTYAMDY (SEQ ID NO: 12) or
- HVR-Ll comprising RASES VXiGYGX 2 SFLH (SEQ ID NO:4), wherein X l is D or S, and X 2 is N or Y;
- HVR-L2 comprising LASNLNS (SEQ ID NO:5);
- HVR-L3 comprising QQNNVDPWT (SEQ ID NO:6)
- an anti-HCV antibody of the invention comprises a heavy chain variable region and a light chain variable region
- HVR-Hl comprising GXiSX 2 TSGYWN (SEQ ID NO: l), wherein X l is D or E, and X 2 is I or L;
- HVR-H2 comprising YISYSGSTYYXiX 2 SLRS (SEQ ID NO:2), wherein Xi is N or S, and wherein X 2 is P or L;
- HVR-H3 comprising ALITTX ⁇ TY AMD Y (SEQ ID NO:3), wherein ⁇ is S or T;
- HVR-L1 comprising RASES VDGYGYSFLH (SEQ ID NO: 14),
- RASES VDGYGNSFLH (SEQ ID NO: 15), or RASESVSGYGYSFLH (SEQ ID NO: 16)
- HVR-L2 comprising LASNLNS (SEQ ID NO:5);
- HVR-L3 comprising QQNNVDPWT (SEQ ID NO:6)
- the invention provides an anti-HCV antibody comprising (a) HVR-Hl comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NOs:9 or 10; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 12; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 14; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the invention provides an anti-HCV antibody comprising (a) HVR-Hl comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NOs:9 or 10; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 15; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the invention provides an anti-HCV antibody comprising (a) HVR-Hl comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 11 ; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 12; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NOs: 14 or 15; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the invention provides an anti-HCV antibody comprising (a) HVR-Hl comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 11 ; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 14; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the invention provides an anti-HCV antibody comprising (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 8; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:9; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 16; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- an anti-HCV antibody of the invention comprises a heavy chain variable region sequence of
- DIVLTQSPSSLSASVGDRVTITCRASESVDGYGYSFLHWFQQKPGKAPKLLIYLASNL NSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQNNVDPWTFGQGTKLEIK SEQ ID NO:24
- DIVLTQSPSSLSASVGDRVTITCRASESVDGYGNSFLHWFQQKPGKAPKLLIYLASNL NSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQNNVDPWTFGQGTKLEIK SEQ ID NO:25
- DIVLTQSPSSLSASVGDRVTITCRASESVSGYGYSFLHWFQQKPGKAPKLLIYLASNL NSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQNNVDPWTFGQGTKLEIK (SEQ ID NO:26).
- an anti-HCV antibody of the invention is not an antibody comprising a heavy chain variable region sequence of
- DIVLTQSPSSLSASVGDRVTITCRASESVDGYGNSFLHWFQQKPGKAPKLLIYLASNL NSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQNNVDPWTFGQGTKLEIK (SEQ ID NO:25).
- an anti-HCV antibody of the invention comprises a heavy chain sequence of
- the invention also provides anti-HCV antibodies comprising three CDRs of the heavy chain and three CDRs of the light chain of anti-HCV antibodies described herein (e.g., V361, V362, VI, V6, V79, V317, V335, V355).
- the invention also provides anti-HCV antibodies comprising a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence SGYWN (SEQ ID NO:33), CDR H2 comprising the amino acid sequence of SEQ ID NO:9, 10, or 11, CDR H3 comprising the amino acid sequence ITTSTYAMDY (SEQ ID NO:34) or ITTTTYAMDY (SEQ ID NO:35); and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, 15, or 16, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- the heavy chain variable region comprises CDR HI comprising the amino acid sequence SGYWN (SEQ ID NO:33), CDR H2 comprising the amino acid sequence of SEQ ID NO:9, 10, or 11, CDR H3 comprising the amino acid sequence ITTSTYAMDY (SEQ ID NO:34) or ITTT
- the anti-HCV antibody is not an antibody comprising a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR
- the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 10, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 10, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the
- the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO:9, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO:9, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 compris
- the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the
- the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:34; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:34; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the
- the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO:9, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 16, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO:9, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 16, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 compris
- the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:34; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
- an anti-HCV antibody is humanized.
- an anti-HCV antibody comprises HVRs as in any of the above embodiments, and further comprises an acceptor human framework, e.g. a human immunoglobulin framework or a human consensus framework.
- an anti-HCV antibody comprises HVRs as in any of the above embodiments, and further comprises a VL comprising an FR1, FR2, FR3, or FR4 sequence as shown in Figure 2A.
- an anti-HCV antibody comprises HVRs as in any of the above embodiments, and further comprises a VH comprising an FR1, FR2, FR3, or FR4 sequence as shown in Figure 2B.
- an anti-HCV antibody comprises a heavy chain variable domain (VH) sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to the amino acid sequence of SEQ ID NOs: 17, 18, 19, 20, 21, 22, or 23.
- VH heavy chain variable domain
- a VH sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity contains substitutions (e.g., conservative
- substitutions substitutions), insertions, or deletions relative to the reference sequence, but an anti-HCV antibody comprising that sequence retains the ability to bind to HCV E2 protein.
- a total of 1 to 10 amino acids have been substituted, inserted and/or deleted in SEQ ID NOs: 17, 18, 19, 20, 21, 22, or 23.
- substitutions, insertions, or deletions occur in regions outside the HVRs (i.e., in the FRs).
- the anti-HCV antibody comprises the VH sequence in SEQ ID NOs: 17, 18, 19, 20, 21, 22, or 23, including post-translational modifications of that sequence.
- the VH comprises one, two or three HVRs selected from: (a) HVR-H1 comprising the amino acid sequence of SEQ ID NOs:7 or 8, (b) HVR-H2 comprising the amino acid sequence of SEQ ID NOs:9, 10, or 11, and (c) HVR-H3 comprising the amino acid sequence of SEQ ID NOs: 12 or 13.
- the VH is not a VH comprising three HVRs consisting of: (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:7, (b) HVR- H2 comprising the amino acid sequence of SEQ ID NO: 11, and (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13.
- an anti-HCV antibody comprising a light chain variable domain (VL) having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to the amino acid sequence of SEQ ID NOs:24, 25, or 26.
- VL light chain variable domain
- a VL sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity contains substitutions (e.g., conservative substitutions), insertions, or deletions relative to the reference sequence, but an anti-HCV antibody comprising that sequence retains the ability to bind to HCV E2 protein.
- the anti-HCV antibody comprises the VL sequence in SEQ ID NOs:24, 25, or 26, including post- translational modifications of that sequence.
- the VL comprises one, two or three HVRs selected from (a) HVR-L1 comprising the amino acid sequence of SEQ ID NOs: 14, 15, or 16; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (c) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the VL is not a VL comprising three HVRs consisting of: (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 15; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (c) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- an anti-HCV antibody comprising a VH as in any of the embodiments provided above, and a VL as in any of the embodiments provided above.
- the antibody comprises the VH and VL sequences of SEQ ID NO: 17 and SEQ ID NO:24, respectively, including post-translational modifications of those sequences.
- the antibody comprises the VH and VL sequences of SEQ ID NO: 18 and SEQ ID NO:24, respectively, including post- translational modifications of those sequences.
- the antibody comprises the VH and VL sequences of SEQ ID NO: 19 and SEQ ID NO:25, respectively, including post-translational modifications of those sequences.
- the antibody comprises the VH and VL sequences of SEQ ID NO:20 and SEQ ID NO:25, respectively, including post-translational modifications of those sequences.
- the antibody comprises the VH and VL sequences of SEQ ID NO:21 and SEQ ID NO:24, respectively, including post-translational modifications of those sequences.
- the antibody comprises the VH and VL sequences of SEQ ID NO:22 and SEQ ID NO:24, respectively, including post-translational modifications of those sequences.
- the antibody comprises the VH and VL sequences of SEQ ID NO:23 and SEQ ID NO:26, respectively, including post-translational modifications of those sequences.
- the antibody comprises the VH and VL sequences of SEQ ID NO:22 and SEQ ID NO:25, respectively, including post-translational modifications of those sequences.
- the invention provides an antibody that binds to the same epitope as an anti-HCV antibody provided herein.
- an anti- HCV antibody is provided that binds to the same epitope as an anti-HCV antibody
- an anti-HCV antibody that binds to an epitope within a fragment of HCV E2 protein consisting of the amino acid sequence of QLINTNGSWHIN (SEQ ID NO:32).
- an anti-HCV antibody is provided that binds to an epitope within a fragment of HCV E2 protein consisting of the amino acid sequence of QLINTNGSWHIN (SEQ ID NO:32) or a homologous amino acid sequence isolated from an HCV genotype selected from the group consisting of genotype 1 (e.g., genotype la and genotype lb), genotype 2 (e.g.
- an anti-HCV antibody that binds to an epitope within a fragment of an HCV E2 protein consisting of an amino acid sequence similar to the sequence of amino acids 412 to 423 of an HCV E2 protein isolated from HCV genotype 1.
- the term "homologous amino acid sequence” refers to an epitope that has a similar amino acid sequence to the amino acid sequence of QLINTNGSWHIN (SEQ ID NO:32) found in genotype la or genotype lb HCV E2 protein.
- the HCV E2 protein is soluble HCV E2 protein.
- the HCV E2 protein is an HCV E2 protein fragment (such as amino acids 1 to 661 of HCV E2 protein isolated from HCV genotype la). Exemplary HCV E2 proteins can be found in, but not limited to, Bukh J et al., Semin Liv Dis., (1995)., 15(l):41-63.
- an anti-HCV antibody is a monoclonal antibody, including a chimeric, humanized or human antibody.
- an anti-HCV antibody is an antibody fragment, e.g., a Fv, Fab, Fab'-SH, scFv, diabody, or F(ab') 2 fragment.
- the antibody is a full length antibody, e.g., an intact IgGl antibody or other antibody class or isotype as defined herein.
- an anti-HCV antibody according to any of the above
- an anti-HCV antibody provided herein has a dissociation constant (Kd) of ⁇ ⁇ , ⁇ 100 nM, ⁇ 10 nM, ⁇ 1 nM, ⁇ 0.1 nM, ⁇ 0.01 nM, or ⁇ 0.001 nM (e.g. 10 ⁇ 8 M or less, e.g. from 10 ⁇ 8 M to 10 "13 M, e.g., from 10 "9 M to 10 "13 M).
- Kd dissociation constant
- an anti-HCV antibody provided herein has a Kd for a binding partner (such as HCV E2 protein or fragment thereof) of less than about any of about 1.0 mM, 500 ⁇ , 100 ⁇ , 50 ⁇ , 25 ⁇ , 10 ⁇ , 5 ⁇ , 1 ⁇ , 900 ⁇ , 800 ⁇ , 700 ⁇ , 600 ⁇ , 500 ⁇ , 400 ⁇ , 350 ⁇ , 300 ⁇ , 250 ⁇ , 200 ⁇ , 150 ⁇ , 100 ⁇ ,95 ⁇ , 90 ⁇ , 85 ⁇ , 80 ⁇ , 75 ⁇ , 70 ⁇ , 65 ⁇ , 60 ⁇ , 55 ⁇ , 50 ⁇ , 45 ⁇ , 40 ⁇ , 35 ⁇ , 30 ⁇ , 25 ⁇ , 20 ⁇ , 15 ⁇ , 10 ⁇ , 5 nM, 1 ⁇ , 900 ⁇ , 800 ⁇ , 700 ⁇ , 600 ⁇ , 500 ⁇ , 400 ⁇ , 300 ⁇ , 200 ⁇ , 100 ⁇ , 50 ⁇ , 25 ⁇ , 12.5
- an anti-HCV antibody described herein binds to HCV E2 protein or fragment thereof with a higher affinity compared to the binding of a parent antibody (such as MRCT10.1) to HCV E2 protein or fragment thereof.
- a parent antibody such as MRCT10.1
- anti-HCV antibody binds to HCV E2 protein or a fragment thereof with at least any of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 ,17, 18, 19, or 20, inclusive, including any value in between these numbers, higher fold affinity (or lowed K D ) compared to the binding of the parent antibody (such as MRCT10.1) to the HCV E2 protein or fragment thereof.
- an anti-HCV antibody described herein demonstrates a similar binding affinity to HCV E2 protein or fragment thereof as compared to the binding affinity of a parent antibody (such as MRCT10.1) to HCV E2 protein or fragment thereof.
- the anti-HCV antibody has a higher HCV inhibitory potency (such as lower EC90) as compared to the HCV inhibitory potency of a parent antibody (such as MRCT10.1).
- Kd is measured by a radiolabeled antigen binding assay (RIA) performed with the Fab version of an antibody of interest and its antigen as described by the following assay.
- RIA radiolabeled antigen binding assay
- Fabs for antigen is measured by equilibrating Fab with a minimal concentration of ( 125 I)- labeled antigen in the presence of a titration series of unlabeled antigen, then capturing bound antigen with an anti-Fab antibody-coated plate (see, e.g., Chen et al., J. Mol. Biol. 293:865- 881(1999)).
- the Fab of interest is then incubated overnight; however, the incubation may continue for a longer period (e.g., about 65 hours) to ensure that equilibrium is reached. Thereafter, the mixtures are transferred to the capture plate for incubation at room temperature (e.g., for one hour). The solution is then removed and the plate washed eight times with 0.1% polysorbate 20 (TWEEN-20 ® ) in PBS. When the plates have dried, 150 ⁇ /well of scintillant (MICROSCINT-20TM; Packard) is added, and the plates are counted on a TOPCOUNTTM gamma counter (Packard) for ten minutes. Concentrations of each Fab that give less than or equal to 20% of maximal binding are chosen for use in competitive binding assays.
- Kd is measured using surface plasmon resonance assays using a BIACORE ® -2000 or a BIACORE ® -3000 (BIAcore, Inc.,
- CM5 chips Piscataway, NJ
- immobilized antigen CM5 chips at -10 response units (RU).
- carboxymethylated dextran biosensor chips CM5, BIACORE, Inc.
- EDC N-ethyl-N'- (3-dimethylaminopropyl)-carbodiimide hydrochloride
- NHS N- hydroxysuccinimide
- Antigen is diluted with 10 mM sodium acetate, pH 4.8, to 5 ⁇ g/ml (-0.2 ⁇ ) before injection at a flow rate of 5 ⁇ /minute to achieve approximately 10 response units (RU) of coupled protein.
- the Kd is measure as described in Example 2.
- an antibody provided herein is an antibody fragment.
- Antibody fragments include, but are not limited to, Fab, Fab', Fab'-SH, F(ab') 2 , Fv, and scFv fragments, and other fragments described below.
- Fab fragment antigen
- Fab' fragment antigen binding domain
- Fab'-SH fragment antigen binding domain antigen binding domain antigen binding domain antigen binding domain antigen binding domain antigen binding domains
- Fv fragment antigen binding domain antigen binding
- scFv fragments see, e.g., Pluckthiin, in The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., (Springer- Verlag, New York), pp. 269-315 (1994); see also WO 93/16185; and U.S.
- Patent Nos. 5,571,894 and 5,587,458 For discussion of Fab and F(ab') 2 fragments comprising salvage receptor binding epitope residues and having increased in vivo half-life, see U.S. Patent No. 5,869,046.
- Diabodies are antibody fragments with two antigen-binding sites that may be bivalent or bispecific. See, for example, EP 404,097; WO 1993/01161; Hudson et al., Nat. Med. 9: 129-134 (2003); and Hollinger et al., Proc. Natl. Acad. Sci. USA 90: 6444-6448 (1993). Triabodies and tetrabodies are also described in Hudson et al., Nat. Med. 9: 129-134 (2003).
- Single-domain antibodies are antibody fragments comprising all or a portion of the heavy chain variable domain or all or a portion of the light chain variable domain of an antibody.
- a single-domain antibody is a human single-domain antibody (Domantis, Inc., Waltham, MA; see, e.g., U.S. Patent No. 6,248,516 Bl).
- Antibody fragments can be made by various techniques, including but not limited to proteolytic digestion of an intact antibody as well as production by recombinant host cells (e.g. E. coli or phage), as described herein.
- recombinant host cells e.g. E. coli or phage
- an antibody provided herein is a chimeric antibody.
- chimeric antibodies are described, e.g., in U.S. Patent No. 4,816,567; and Morrison et al., Proc. Natl. Acad. Sci. USA, 81:6851-6855 (1984)).
- a chimeric antibody comprises a non-human variable region (e.g., a variable region derived from a mouse, rat, hamster, rabbit, or non-human primate, such as a monkey) and a human constant region.
- a chimeric antibody is a "class switched" antibody in which the class or subclass has been changed from that of the parent antibody. Chimeric antibodies include antigen-binding fragments thereof.
- a chimeric antibody is a humanized antibody.
- a non-human antibody is humanized to reduce immunogenicity to humans, while retaining the specificity and affinity of the parental non-human antibody.
- a humanized antibody comprises one or more variable domains in which HVRs, e.g., CDRs, (or portions thereof) are derived from a non-human antibody, and FRs (or portions thereof) are derived from human antibody sequences.
- HVRs e.g., CDRs, (or portions thereof) are derived from a non-human antibody
- FRs or portions thereof
- a humanized antibody optionally will also comprise at least a portion of a human constant region.
- some FR residues in a humanized antibody are substituted with corresponding residues from a non-human antibody (e.g., the antibody from which the HVR residues are derived), e.g., to restore or improve antibody specificity or affinity.
- a non-human antibody e.g., the antibody from which the HVR residues are derived
- Human framework regions that may be used for humanization include but are not limited to: framework regions selected using the "best-fit" method (see, e.g., Sims et al. J. Immunol. 151:2296 (1993)); framework regions derived from the consensus sequence of human antibodies of a particular subgroup of light or heavy chain variable regions (see, e.g., Carter et al. Proc. Natl. Acad. Sci. USA, 89:4285 (1992); and Presta et al. J. Immunol, 151:2623 (1993)); human mature (somatically mutated) framework regions or human germline framework regions (see, e.g., Almagro and Fransson, Front. Biosci.
- an antibody provided herein is a human antibody.
- Human antibodies can be produced using various techniques known in the art. Human antibodies are described generally in van Dijk and van de Winkel, Curr. Opin. Pharmacol. 5: 368-74 (2001) and Lonberg, Curr. Opin. Immunol. 20:450-459 (2008).
- Human antibodies may be prepared by administering an immunogen to a transgenic animal that has been modified to produce intact human antibodies or intact antibodies with human variable regions in response to antigenic challenge. Such animals typically contain all or a portion of the human immunoglobulin loci, which replace the endogenous
- Human antibodies can also be made by hybridoma-based methods. Human myeloma and mouse-human heteromyeloma cell lines for the production of human monoclonal antibodies have been described. (See, e.g., Kozbor J. Immunol., 133: 3001 (1984); Brodeur et al., Monoclonal Antibody Production Techniques and Applications, pp. 51-63 (Marcel Dekker, Inc., New York, 1987); and Boerner et al., J. Immunol., 147: 86 (1991).) Human antibodies generated via human B-cell hybridoma technology are also described in Li et al, Proc. Nad. Acad. Sti.
- an anti-HCV antibody is generated using the method described in Example 1. Techniques for selecting human antibodies from antibody libraries are described below.
- Antibodies of the invention may be isolated by screening combinatorial libraries for antibodies with the desired activity or activities. For example, a variety of methods are known in the art for generating phage display libraries and screening such libraries for antibodies possessing the desired binding characteristics. Such methods are reviewed, e.g., in Hoogenboom et al. in Methods in Molecular Biology 178: 1-37 (O'Brien et al., ed., Human Press, Totowa, NJ, 2001) and further described, e.g., in the McCafferty et al., Nature
- phage display methods repertoires of VH and VL genes are separately cloned by polymerase chain reaction (PCR) and recombined randomly in phage libraries, which can then be screened for antigen-binding phage as described in Winter et al., Ann. Rev. Immunol., 12: 433-455 (1994). Phage typically display antibody fragments, either as single- chain Fv (scFv) fragments or as Fab fragments. Libraries from immunized sources provide high-affinity antibodies to the immunogen without the requirement of constructing hybridomas.
- PCR polymerase chain reaction
- naive repertoire can be cloned (e.g., from human) to provide a single source of antibodies to a wide range of non-self and also self-antigens without any immunization as described by Griffiths et al., EMBO J, 12: 725-734 (1993).
- naive libraries can also be made synthetically by cloning unrearranged V-gene segments from stem cells, and using PCR primers containing random sequence to encode the highly variable CDR3 regions and to accomplish rearrangement in vitro, as described by Hoogenboom and Winter, /. Mol. Biol., 227: 381-388 (1992).
- Patent publications describing human antibody phage libraries include, for example: US Patent No. 5,750,373, and US Patent Publication Nos. 2005/0079574, 2005/0119455, 2005/0266000, 2007/0117126, 2007/0160598,
- Antibodies or antibody fragments isolated from human antibody libraries are considered human antibodies or human antibody fragments herein.
- an antibody provided herein is a multispecific antibody, e.g. a bispecific antibody.
- Multispecific antibodies are monoclonal antibodies that have binding specificities for at least two different sites.
- one of the binding specificities is for HCV E2 protein and the other is for any other antigen.
- bispecific antibodies may bind to two different epitopes of HCV E2 protein.
- Bispecific antibodies may also be used to localize cytotoxic agents to HCV that expresses HCV E2 protein.
- Bispecific antibodies can be prepared as full length antibodies or antibody fragments.
- Multispecific antibodies include, but are not limited to, recombinant co-expression of two immunoglobulin heavy chain-light chain pairs having different specificities (see Milstein and Cuello, Nature 305: 537 (1983)), WO 93/08829, and Traunecker et al., EMBO J. 10: 3655 (1991)), and "knob-in-hole” engineering (see, e.g., U.S. Patent No. 5,731,168). Multi- specific antibodies may also be made by engineering electrostatic steering effects for making antibody Fc-heterodimeric molecules
- Engineered antibodies with three or more functional antigen binding sites are also included herein (see, e.g. US 2006/0025576A1).
- the antibody or fragment herein also includes a "Dual Acting FAb” or “DAF” comprising an antigen binding site that binds to HCV E2 protein as well as another, different antigen (see, US 2008/0069820, for example).
- amino acid sequence variants of the antibodies provided herein are contemplated. For example, it may be desirable to improve the binding affinity and/or other biological properties of the antibody.
- Amino acid sequence variants of an antibody may be prepared by introducing appropriate modifications into the nucleotide sequence encoding the antibody, or by peptide synthesis. Such modifications include, for example, deletions from, and/or insertions into and/or substitutions of residues within the amino acid sequences of the antibody. Any combination of deletion, insertion, and substitution can be made to arrive at the final construct, provided that the final construct possesses the desired characteristics, e.g., antigen-binding. a) Substitution, Insertion, and Deletion Variants
- antibody variants having one or more amino acid substitutions are provided.
- Sites of interest for substitutional mutagenesis include the HVRs and FRs. Conservative substitutions are shown in Table A under the heading of
- amino acid side chain classes “conservative substitutions.” More substantial changes are provided in Table A under the heading of "exemplary substitutions,” and as further described below in reference to amino acid side chain classes. Amino acid substitutions may be introduced into an antibody of interest and the products screened for a desired activity, e.g., retained/improved antigen binding, decreased immunogenicity, or improved ADCC or CDC.
- Amino acids may be grouped according to common side-chain properties:
- Non-conservative substitutions will entail exchanging a member of one of these classes for another class.
- substitutional variant involves substituting one or more hypervariable region residues of a parent antibody (e.g. a humanized or human antibody).
- a parent antibody e.g. a humanized or human antibody
- the resulting variant(s) selected for further study will have modifications (e.g., improvements) in certain biological properties (e.g., increased affinity, reduced immunogenicity) relative to the parent antibody and/or will have substantially retained certain biological properties of the parent antibody.
- An exemplary substitutional variant is an affinity matured antibody, which may be conveniently generated, e.g., using phage display-based affinity maturation techniques such as those described herein. Briefly, one or more HVR residues are mutated and the variant antibodies displayed on phage and screened for a particular biological activity (e.g. binding affinity).
- Alterations may be made in HVRs, e.g., to improve antibody affinity. Such alterations may be made in HVR "hotspots," i.e., residues encoded by codons that undergo mutation at high frequency during the somatic maturation process (see, e.g., Chowdhury, Methods Mol. Biol. 207: 179-196 (2008)), and/or SDRs (a-CDRs), with the resulting variant VH or VL being tested for binding affinity.
- HVR "hotspots” i.e., residues encoded by codons that undergo mutation at high frequency during the somatic maturation process (see, e.g., Chowdhury, Methods Mol. Biol. 207: 179-196 (2008)), and/or SDRs (a-CDRs), with the resulting variant VH or VL being tested for binding affinity.
- Affinity maturation by constructing and reselecting from secondary libraries has been described, e.g., in Hoogenboo
- affinity maturation diversity is introduced into the variable genes chosen for maturation by any of a variety of methods (e.g., error-prone PCR, chain shuffling, or oligonucleotide-directed mutagenesis).
- a secondary library is then created. The library is then screened to identify any antibody variants with the desired affinity.
- Another method to introduce diversity involves HVR-directed approaches, in which several HVR residues (e.g., 4-6 residues at a time) are randomized. HVR residues involved in antigen binding may be specifically identified, e.g., using alanine scanning mutagenesis or modeling. CDR-H3 and CDR-L3 in particular are often targeted.
- substitutions, insertions, or deletions may occur within one or more HVRs so long as such alterations do not substantially reduce the ability of the antibody to bind antigen.
- conservative alterations e.g., conservative substitutions as provided herein
- Such alterations may be outside of HVR "hotspots" or SDRs.
- each HVR either is unaltered, or contains no more than one, two or three amino acid substitutions.
- a useful method for identification of residues or regions of an antibody that may be targeted for mutagenesis is called “alanine scanning mutagenesis" as described by
- a residue or group of target residues e.g., charged residues such as arg, asp, his, lys, and glu
- a neutral or negatively charged amino acid e.g., alanine or polyalanine
- Further substitutions may be introduced at the amino acid locations demonstrating functional sensitivity to the initial substitutions.
- a crystal structure of an antigen-antibody complex to identify contact points between the antibody and antigen. Such contact residues and neighboring residues may be targeted or eliminated as candidates for substitution.
- Variants may be screened to determine whether they contain the desired properties.
- Amino acid sequence insertions include amino- and/or carboxyl-terminal fusions ranging in length from one residue to polypeptides containing a hundred or more residues, as well as intrasequence insertions of single or multiple amino acid residues.
- terminal insertions include an antibody with an N-terminal methionyl residue.
- Other insertional variants of the antibody molecule include the fusion to the N- or C-terminus of the antibody to an enzyme (e.g. for ADEPT) or a polypeptide which increases the serum half-life of the antibody.
- ADEPT enzyme
- an antibody provided herein is altered to increase or decrease the extent to which the antibody is glycosylated.
- Addition or deletion of glycosylation sites to an antibody may be conveniently accomplished by altering the amino acid sequence such that one or more glycosylation sites is created or removed.
- the carbohydrate attached thereto may be altered.
- Native antibodies produced by mammalian cells typically comprise a branched, biantennary oligosaccharide that is generally attached by an N-linkage to Asn297 of the CH2 domain of the Fc region. See, e.g., Wright et al. TIBTECH 15:26-32 (1997).
- oligosaccharide may include various carbohydrates, e.g., mannose, N-acetyl glucosamine (GlcNAc), galactose, and sialic acid, as well as a fucose attached to a GlcNAc in the "stem" of the biantennary oligosaccharide structure.
- modifications of the oligosaccharide in an antibody of the invention may be made in order to create antibody variants with certain improved properties.
- antibody variants having a carbohydrate structure that lacks fucose attached (directly or indirectly) to an Fc region.
- the amount of fucose in such antibody may be from 1% to 80%, from 1% to 65%, from 5% to 65% or from 20% to 40%.
- the amount of fucose is determined by calculating the average amount of fucose within the sugar chain at Asn297, relative to the sum of all glycostructures attached to Asn 297 (e. g. complex, hybrid and high mannose structures) as measured by MALDI-TOF mass spectrometry, as described in WO 2008/077546, for example.
- Asn297 refers to the asparagine residue located at about position 297 in the Fc region (Eu numbering of Fc region residues); however, Asn297 may also be located about + 3 amino acids upstream or downstream of position 297, i.e., between positions 294 and 300, due to minor sequence variations in antibodies. Such fucosylation variants may have improved ADCC function. See, e.g., US Patent Publication Nos. US 2003/0157108 (Presta, L.); US 2004/0093621 (Kyowa Hakko Kogyo Co., Ltd).
- Examples of cell lines capable of producing defucosylated antibodies include Led 3 CHO cells deficient in protein fucosylation (Ripka et al. Arch. Biochem. Biophys.
- knockout cell lines such as alpha- 1,6- fucosyltransferase gene, FUT8, knockout CHO cells (see, e.g., Yamane-Ohnuki et al.
- Antibodies variants are further provided with bisected oligosaccharides, e.g., in which a biantennary oligosaccharide attached to the Fc region of the antibody is bisected by GlcNAc. Such antibody variants may have reduced fucosylation and/or improved ADCC function. Examples of such antibody variants are described, e.g., in WO 2003/011878 (Jean- Mairet et al.); US Patent No. 6,602,684 (Umana et al.); and US 2005/0123546 (Umana et al). Antibody variants with at least one galactose residue in the oligosaccharide attached to the Fc region are also provided.
- Such antibody variants may have improved CDC function.
- Such antibody variants are described, e.g., in WO 1997/30087 (Patel et al.); WO 1998/58964 (Raju, S.); and WO 1999/22764 (Raju, S.).
- an anti-HCV antibody glycosylation variant comprises a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23, and a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25.
- one or more amino acid modifications may be introduced into the Fc region of an antibody provided herein, thereby generating an Fc region variant.
- the Fc region variant may comprise a human Fc region sequence (e.g., a human IgGl, IgG2, IgG3 or IgG4 Fc region) comprising an amino acid modification (e.g. a substitution) at one or more amino acid positions.
- the invention contemplates an antibody variant that possesses some but not all effector functions, which make it a desirable candidate for applications in which the half life of the antibody in vivo is important yet certain effector functions (such as complement and ADCC) are unnecessary or deleterious.
- In vitro and/or in vivo cytotoxicity assays can be conducted to confirm the reduction/depletion of CDC and/or ADCC activities.
- Fc receptor (FcR) binding assays can be conducted to ensure that the antibody lacks FcyR binding (hence likely lacking ADCC activity), but retains FcRn binding ability.
- NK cells express Fc(RIII only, whereas monocytes express Fc(RI, Fc(RII and Fc(RIII.
- FcR expression on hematopoietic cells is summarized in Table 3 on page 464 of Ravetch and Kinet, Annu. Rev. Immunol.
- non-radioactive assays methods may be employed (see, for example, ACTITM non-radioactive cytotoxicity assay for flow cytometry
- PBMC peripheral blood mononuclear cells
- NK Natural Killer
- ADCC activity of the molecule of interest may be assessed in vivo, e.g., in an animal model such as that disclosed in Clynes et al. Proc. Nat'lAcad. Sci. USA 95:652-656 (1998).
- Clq binding assays may also be carried out to confirm that the antibody is unable to bind Clq and hence lacks CDC activity.
- a CDC assay may be performed (see, for example, Gazzano-Santoro et al., J. Immunol. Methods 202: 163 (1996); Cragg, M.S. et al., Blood 101: 1045-1052 (2003); and Cragg, M.S. and M.J. Glennie, Blood 103:2738-2743 (2004)).
- FcRn binding and in vivo clearance/half life determinations can also be performed using methods known in the art (see, e.g., Petkova, S.B. et al., Int'l. Immunol. 18(12): 1759- 1769 (2006)).
- Antibodies with reduced effector function include those with substitution of one or more of Fc region residues 238, 265, 269, 270, 297, 327 and 329 (U.S. Patent No. 6,737,056).
- Fc mutants include Fc mutants with substitutions at two or more of amino acid positions 265, 269, 270, 297 and 327, including the so-called "DANA" Fc mutant with substitution of residues 265 and 297 to alanine (US Patent No. 7,332,581).
- an antibody variant comprises an Fc region with one or more amino acid substitutions which improve ADCC, e.g., substitutions at positions 298, 333, and/or 334 of the Fc region (EU numbering of residues).
- alterations are made in the Fc region that result in altered (i.e., either improved or diminished) Clq binding and/or Complement Dependent
- Cytotoxicity e.g., as described in US Patent No. 6,194,551, WO 99/51642, and Idusogie et al. J. Immunol. 164: 4178-4184 (2000).
- Such Fc variants include those with substitutions at one or more of Fc region residues: 238, 256, 265, 272, 286, 303, 305, 307, 311, 312, 317, 340, 356, 360, 362, 376, 378, 380, 382, 413, 424 or 434, e.g., substitution of Fc region residue 434 (US Patent No. 7,371,826).
- cysteine engineered antibodies e.g., "thioMAbs”
- one or more residues of an antibody are substituted with cysteine residues.
- the substituted residues occur at accessible sites of the antibody.
- reactive thiol groups are thereby positioned at accessible sites of the antibody and may be used to conjugate the antibody to other moieties, such as drug moieties or linker-drug moieties, to create an immunoconjugate, as described further herein.
- any one or more of the following residues may be substituted with cysteine: V205 (Kabat numbering) of the light chain; Al 18 (EU numbering) of the heavy chain; and S400 (EU numbering) of the heavy chain Fc region.
- Cysteine engineered antibodies may be generated as described, e.g., in U.S. Patent No. 7,521,541.
- an antibody provided herein may be further modified to contain additional nonproteinaceous moieties that are known in the art and readily available.
- the moieties suitable for derivatization of the antibody include but are not limited to water soluble polymers.
- water soluble polymers include, but are not limited to, polyethylene glycol (PEG), copolymers of ethylene glycol/propylene glycol, carboxymethylcellulose, dextran, polyvinyl alcohol, polyvinyl pyrrolidone, poly-1, 3- dioxolane, poly-1, 3, 6-trioxane, ethylene/maleic anhydride copolymer, polyaminoacids (either homopolymers or random copolymers), and dextran or poly(n-vinyl pyrrolidone)polyethylene glycol, propropylene glycol homopolymers, prolypropylene oxide/ethylene oxide copolymers, polyoxyethylated polyols (e.g., g
- Polyethylene glycol propionaldehyde may have advantages in manufacturing due to its stability in water.
- the polymer may be of any molecular weight, and may be branched or unbranched.
- the number of polymers attached to the antibody may vary, and if more than one polymer are attached, they can be the same or different molecules. In general, the number and/or type of polymers used for derivatization can be determined based on considerations including, but not limited to, the particular properties or functions of the antibody to be improved, whether the antibody derivative will be used in a therapy under defined conditions, etc.
- conjugates of an antibody and nonproteinaceous moiety that may be selectively heated by exposure to radiation are provided.
- the nonproteinaceous moiety is a carbon nanotube (Kam et al., Proc. Natl. Acad. Sci. USA 102: 11600-11605 (2005)).
- the radiation may be of any wavelength, and includes, but is not limited to, wavelengths that do not harm ordinary cells, but which heat the nonproteinaceous moiety to a temperature at which cells proximal to the antibody-nonproteinaceous moiety are killed.
- polypeptides can be prepared using isolated nucleic acids encoding such antibodies or fragments thereof, vectors and host-cells comprising such nucleic acids.
- Antibodies may be produced using recombinant methods and compositions, e.g., as described in U.S. Patent No. 4,816,567.
- isolated nucleic acid encoding an anti-HCV antibody described herein is provided. Such nucleic acid may encode an amino acid sequence comprising the VL and/or an amino acid sequence comprising the VH of the antibody (e.g., the light and/or heavy chains of the antibody).
- the isolated nucleic acid encodes a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23.
- the isolated nucleic acid encodes a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25.
- the isolated nucleic acid encodes a heavy chain amino acid sequence selected from the group consisting of SEQ ID NOs:27 and 28. In some embodiments, the isolated nucleic acid encodes a light chain amino acid sequence of SEQ ID NO:30.
- nucleic acids encoding the desired antibodies or antibody fragments as described herein are isolated and inserted into a replicable vector for further cloning (amplification of the DNA) or for expression.
- one or more vectors comprising such nucleic acid are provided.
- a vector comprises a nucleic acid encoding a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23.
- a vector comprises a nucleic acid encoding a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25.
- a vector comprises a nucleic acid encoding a heavy chain amino acid sequence selected from the group consisting of SEQ ID NOs:27 and 28. In still other embodiments, a vector comprises a nucleic acid encoding a light chain amino acid sequence of SEQ ID NO:30.
- DNA encoding the polyclonal or monoclonal antibodies is readily isolated (e.g., with oligonucleotide probes that specifically bind to genes encoding the heavy and light chains of the antibody) and sequenced using conventional procedures. Many cloning and/or expression vectors are commercially available.
- Vector components generally include, but are not limited to, one or more of the following, a signal sequence, an origin of replication, one or more marker genes, a multiple cloning site containing recognition sequences for numerous restriction endonucleases, an enhancer element, a promoter, and a transcription termination sequence.
- the antibodies or fragments thereof may be produced recombinantly not only directly, but also as a fusion protein, where the antibody is fused to a heterologous polypeptide, preferably a signal sequence or other polypeptide having a specific cleavage site at the N-terminus of the mature protein or polypeptide.
- a heterologous polypeptide preferably a signal sequence or other polypeptide having a specific cleavage site at the N-terminus of the mature protein or polypeptide.
- the heterologous signal sequence selected preferably is one that is recognized and processed (i.e., cleaved by a signal peptidase) by eukaryotic host-cells.
- the eukaryotic (i.e., mammalian) signal sequence is replaced by a prokaryotic signal sequence selected, for example, from the group consisting of leader sequences from alkaline phosphatase, penicillinase, lpp, or heat-stable enterotoxin II genes.
- a prokaryotic signal sequence selected, for example, from the group consisting of leader sequences from alkaline phosphatase, penicillinase, lpp, or heat-stable enterotoxin II genes.
- yeast secretion the native signal sequence may be substituted by, e.g., the yeast invertase leader, factor leader (including Saccharomyces and Kluyveromyces - factor leaders), or acid phosphatase leader, the C. albicans glucoamylase leader, or the signal described in WO 90/13646.
- mammalian signal sequences as well as viral secretory leaders for example, the herpes simplex virus gD signal,
- the DNA for such precursor region is ligated in reading frame to the DNA encoding the antibodies or fragments thereof.
- Both expression and cloning vectors contain a nucleic acid sequence that enables the vector to replicate in one or more selected host-cells.
- this sequence is one that enables the vector to replicate independently of the host chromosomal DNA, and includes origins of replication or autonomously replicating sequences.
- Such sequences are well known for a variety of bacteria, yeast, and viruses.
- the origin of replication from the plasmid pBR322 is suitable for most Gram-negative bacteria, the 2 ⁇ plasmid origin is suitable for yeast, and various viral origins (SV40, polyoma, adenovirus, vesicular stomatitis virus (“VSV”) or bovine papilloma virus (“BPV”) are useful for cloning vectors in mammalian cells.
- viral origins SV40, polyoma, adenovirus, vesicular stomatitis virus (“VSV”) or bovine papilloma virus (“BPV) are useful for cloning vectors in mammalian cells.
- VSV vesicular stomatitis virus
- BBV bovine papilloma virus
- the origin of replication component is not needed for mammalian expression vectors (the SV40 origin may typically be used only because it contains the early promoter).
- Expression and cloning vectors may also contain a selection gene, known as a selectable marker.
- selection genes encode proteins that (a) confer resistance to antibiotics or other toxins, e.g., ampicillin, neomycin, methotrexate, or tetracycline, (b) complement auxotrophic deficiencies, or (c) supply critical nutrients not available from complex media, e.g., the gene encoding D-alanine racemase for Bacilli.
- One example of a selection scheme utilizes a drug to arrest growth of a host-cell. Those cells that are successfully transformed with a heterologous gene produce a protein conferring drug resistance and thus survive the selection regimen. Examples of such dominant selection strategies use the drugs neomycin, mycophenolic acid and hygromycin.
- DHFR dihydrofolate reductase
- thymidine kinase metallothionein-I and -II
- primate metallothionein genes adenosine deaminase, ornithine decarboxylase, and the like.
- Mtx methotrexate
- An exemplary host-cell strain for use with wild-type DHFR is the Chinese hamster ovary (“CHO”) cell line lacking DHFR activity (e.g., ATCC CRL-9096).
- Suitable selectable markers for mammalian cells are those that enable the identification of cells competent to take up the antibody- or antibody fragment- encoding nucleic acids, such as dihydrofolate reductase ("DHFR"), glutamine synthetase (GS), thymidine kinase, metallothionein-I and -II, preferably primate metallothionein genes, adenosine deaminase, ornithine decarboxylase, and the like.
- DHFR dihydrofolate reductase
- GS glutamine synthetase
- thymidine kinase metallothionein-I and -II
- metallothionein-I and -II preferably primate metallothionein genes, adenosine deaminase, ornithine decarboxylase, and the like.
- GS glucose synthetase
- Msx L-methionine sulfoximine
- the GS selection/amplification system may be used in combination with the DHFR selection/amplification system described above.
- host-cells (particularly wild-type hosts that contain endogenous DHFR) transformed or co-transformed with DNA sequences encoding anti-CD83 agonist antibodies or fragments thereof, wild-type DHFR protein, and another selectable marker such as aminoglycoside 3'-phosphotransferase ("APH") can be selected by cell growth in medium containing a selection agent for the appropriate selectable marker, such as an aminoglycosidic antibiotic, e.g., kanamycin, neomycin, or G418. See U.S. Patent No. 4,965,199.
- APH aminoglycoside 3'-phosphotransferase
- a suitable selection gene for use in yeast is the trpl gene present in the yeast plasmid YRp7 (Stinchcomb et al., Nature, 282:39 (1979)).
- the trpl gene provides a selection marker for a mutant strain of yeast lacking the ability to grow medium containing tryptophan (e.g., ATCC No. 44076 or PEP4-1). Jones, Genetics, 85: 12 (1977).
- the presence of the trpl lesion in the yeast host-cell genome then provides an effective environment for detecting transformation by growth in the absence of tryptophan.
- Lew2-deficient yeast strains e.g., ATCC 20,622 or 38,626) can be complemented by known plasmids bearing the Leu2 gene.
- vectors derived from the 1.6 ⁇ circular plasmid pKDl can be used for transformation of Kluyveromyces yeasts.
- an expression system for large-scale production of recombinant calf chymosin was reported for K. lactis. Van den Berg,
- Expression and cloning vectors usually contain a promoter that is recognized by the host organism and is operably linked to the nucleic acid encoding the anti-HCV antibodies or fragments thereof.
- Promoters suitable for use with prokaryotic hosts include the phoA promoter, lactamase and lactose promoter systems, alkaline phosphatase promoter, a tryptophan promoter system, and hybrid promoters such as the tac promoter, although other known bacterial promoters are also suitable. Promoters for use in bacterial systems also will contain a Shine-Dalgarno (S.D.) sequence operably linked to the DNA encoding the antibodies and antibody fragments.
- S.D. Shine-Dalgarno
- Promoter sequences are known for eukaryotes. Virtually all eukaryotic genes have an AT-rich region located approximately 25 to 30 bases upstream from the site where transcription is initiated. Another sequence found 70 to 80 bases upstream from the start of transcription of many genes is a CNCAAT region where N may be any nucleotide. At the 3' end of most eukaryotic genes is an AATAAA sequence that may be the signal for addition of the polyA tail to the 3' end of the coding sequence. All of these sequences may be inserted into eukaryotic expression vectors.
- suitable promoter sequences for use with yeast hosts include the promoters for 3-phosphoglycerate kinase or other glycolytic enzymes, such as enolase, glyceraldehyde-3-phosphate dehydrogenase, hexokinase, pyruvate decarboxylase, phospho-fructokinase, glucose-6-phosphate isomerase, 3-phosphoglycerate mutase, pyruvate kinase, triosephosphate isomerase, phosphoglucose isomerase, and glucokinase.
- 3-phosphoglycerate kinase or other glycolytic enzymes such as enolase, glyceraldehyde-3-phosphate dehydrogenase, hexokinase, pyruvate decarboxylase, phospho-fructokinase, glucose-6-phosphate isomerase, 3-phosphoglycerate mutase, pyruv
- inducible promoters in yeast have the additional advantage of permitting
- exemplary inducible promoters include the promoter regions for alcohol dehydrogenase 2, isocytochrome C, acid phosphatase, degradative enzymes associated with nitrogen metabolism, metallothionein, glyceraldehyde- 3-phosphate dehydrogenase, and enzymes responsible for maltose and galactose utilization.
- Suitable vectors and promoters for use in yeast expression are further described in EP 73,657.
- Yeast enhancers also are advantageously used with yeast promoters.
- Transcription of nucleic acids encoding antibodies or fragments thereof from vectors in mammalian host-cells can be controlled, for example, by promoters obtained from the genomes of viruses such as polyoma virus, fowlpox virus, adenovirus (such as
- Adenovirus 2 bovine papilloma virus, avian sarcoma virus, cytomegalovirus, a retrovirus, hepatitis-B virus and most preferably Simian Virus 40 (SV40), by heterologous mammalian promoters, e.g., the actin promoter or an immunoglobulin promoter, and by heat-shock gene promoters, provided such promoters are compatible with the desired host-cell systems.
- heterologous mammalian promoters e.g., the actin promoter or an immunoglobulin promoter
- heat-shock gene promoters e.g., SV40
- the early and late promoters of the SV40 virus are conveniently obtained as an SV40 restriction fragment that also contains the SV40 viral origin of replication.
- the immediate early promoter of the human cytomegalovirus is conveniently obtained as a Hindlll E restriction fragment.
- a system for expressing DNA in mammalian hosts using the bovine papilloma virus as a vector is disclosed in U.S. Patent No. 4,419,446. A modification of this system is described in U.S. Patent No. 4,601,978. See also Reyes et al., Nature 297:598-601 (1982), regarding methods for expression of human interferon cDNA in mouse cells under the control of a thymidine kinase promoter from herpes simplex virus.
- Rous Sarcoma Virus long terminal repeat can be used as the promoter.
- Enhancer sequences are now known from mammalian genes (globin, elastase, albumin, a- fetoprotein, and insulin). Typically, however, one of ordinary skill in the art will use an enhancer from a eukaryotic virus. Examples include the SV40 enhancer on the late side of the replication origin (bp 100-270), the cytomegalovirus early promoter enhancer, the polyoma enhancer on the late side of the replication origin, and adenovirus enhancers.
- the enhancer may be spliced into the vector at a position 5' or 3' to the antibody- or antibody-fragment encoding sequences, but is preferably located at a site 5' of the promoter.
- Expression vectors used in eukaryotic host-cells will also contain sequences necessary for the termination of transcription and for stabilizing the mRNA. Such sequences are commonly available from the 5' and, occasionally 3', untranslated regions of eukaryotic or viral DNAs or cDNAs. These regions contain nucleotide segments transcribed as
- One useful transcription termination component is the bovine growth hormone polyadenylation region. See W094/11026 and the expression vector disclosed therein. Selection and transformation of host-cells
- Suitable host cells for cloning or expressing nucleic acid encoding anti-HCV antibodies or fragments thereof in the vectors described include prokaryotic or eukaryotic cells described herein.
- antibodies may be produced in bacteria, in particular when glycosylation and Fc effector function are not needed.
- U.S. Patent Nos. 5,648,237, 5,789,199, and 5,840,523. See also Charlton, Methods in Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ, 2003), pp. 245-254, describing expression of antibody fragments in E. coli.)
- the antibody may be isolated from the bacterial cell paste in a soluble fraction and can be further purified.
- eukaryotic microbes such as filamentous fungi or yeast are suitable cloning or expression hosts for antibody-encoding vectors, including fungi and yeast strains whose glycosylation pathways have been "humanized,” resulting in the production of an antibody with a partially or fully human glycosylation pattern. See
- Suitable host cells for the expression of glycosylated antibody are also derived from multicellular organisms (invertebrates and vertebrates). Examples of invertebrate cells include plant and insect cells. Numerous baculoviral strains have been identified which may be used in conjunction with insect cells, particularly for transfection of Spodoptera frugiperda cells.
- Plant cell cultures can also be utilized as hosts. See, e.g., US Patent Nos. 5,959,177, 6,040,498, 6,420,548, 7,125,978, and 6,417,429 (describing PLANTIBODIESTM technology for producing antibodies in transgenic plants).
- Vertebrate cells may also be used as hosts.
- mammalian cell lines that are adapted to grow in suspension may be useful.
- useful mammalian host cell lines are monkey kidney CV1 line transformed by SV40 (COS-7); human embryonic kidney line (293 or 293 cells as described, e.g., in Graham et al., /. Gen Virol. 36:59 (1977)); baby hamster kidney cells (BHK); mouse Sertoli cells (TM4 cells as described, e.g., in Mather, Biol. Reprod.
- monkey kidney cells (CV1); African green monkey kidney cells (VERO-76); human cervical carcinoma cells (HELA); canine kidney cells (MDCK; buffalo rat liver cells (BRL 3A); human lung cells (W138); human liver cells (Hep G2); mouse mammary tumor (MMT 060562); TRI cells, as described, e.g., in Mather et al., Annals N.Y. Acad. Sci. 383:44-68 (1982); MRC 5 cells; and FS4 cells.
- Other useful mammalian host cell lines include Chinese hamster ovary (CHO) cells, including DHFR " CHO cells (Urlaub et al, Pwc. Natl. Acad. Sci.
- host-cells are transformed with the above-described expression or cloning vectors for anti-HCV antibody or antibody fragment production are cultured in conventional nutrient media modified as appropriate for inducing promoters, selecting transformants, or amplifying the genes encoding the desired sequences.
- Patent Nos. 4,767,704; 4,657,866; 4,927,762; 4,560,655; or 5,122,469; WIPO Publication Nos. WO 90/03430; WO 87/00195; or U.S. Patent Re. 30,985 may be used as culture media for the host-cells.
- any of these media may be supplemented as necessary with hormones and/or other growth factors (such as insulin, transferrin, or epidermal growth factor), salts (such as sodium chloride, calcium, magnesium, and phosphate), buffers (such as HEPES), nucleotides (such as adenosine and thymidine), antibiotics (such as GENTAMYCINTM drug), trace elements (defined as inorganic compounds usually present at final concentrations in the micromolar range), and glucose or an equivalent energy source. Any other necessary supplements may also be included at appropriate concentrations that would be known to those skilled in the art.
- the culture conditions such as temperature, pH, and the like, are those previously used with the host-cell selected for expression, and will be apparent to the ordinarily skilled artisan.
- a host cell comprising one or more nucleic acid encoding an anti-HCV antibody or fragment thereof as described herein.
- a host cell comprises (e.g., has been transformed with): (1) a vector comprising a nucleic acid that encodes an amino acid sequence comprising the VL of the antibody and an amino acid sequence comprising the VH of the antibody, or (2) a first vector comprising a nucleic acid that encodes an amino acid sequence comprising the VL of the antibody and a second vector comprising a nucleic acid that encodes an amino acid sequence comprising the VH of the antibody.
- the host cell is eukaryotic, e.g.
- a host cell comprises a nucleic acid encoding a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23. In other embodiments, a host cell comprises a nucleic acid encoding a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25. In other embodiments, a host cell comprises a nucleic acid encoding a heavy chain amino acid sequence selected from the group consisting of SEQ ID NOs:27 and 28.
- a host cell comprises a nucleic acid encoding a light chain amino acid sequence of SEQ ID NO:30.
- a method of making an anti-HCV antibody comprises culturing a host cell comprising a nucleic acid encoding the antibody, as provided above, under conditions suitable for expression of the antibody, and optionally recovering the antibody from the host cell (or host cell culture medium).
- the host cell is a 293T cell.
- an antibody that binds HCV E2 protein is produced by a method comprising culturing a host cell comprising one or more nucleic acid encoding an antibody described herein, under a condition suitable for expression of the one or more nucleic acid, and recovering the antibody produced by the cell.
- the one or more nucleic acid encodes a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23.
- the one or more nucleic acid encodes a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25.
- the one or more nucleic acid encodes a heavy chain amino acid sequence selected from the group consisting of SEQ ID NOs:27 and 28. In still another further embodiment, the one or more nucleic acid encodes a light chain amino acid sequence of SEQ ID NO:30.
- the antibody that binds HCV E2 protein produced by a method comprising culturing a host cell comprising one or more nucleic acid encoding an antibody described herein has a lysine residue removed from the C- terminus. In some embodiments, the host cell is a 293T cell.
- the anti-HCV antibodies or antibody fragments can be produced intracellularly, in the periplasmic space, or secreted directly into the medium. If the antibodies are produced intracellularly, as a first step, the particulate debris from either host-cells or lysed fragments is removed, for example, by centrifugation or ultrafiltration. Carter et al., Bio/Technology 10: 163-167 (1992) describe a procedure for isolating antibodies which are secreted to the periplasmic space of E. coli.
- cell paste is thawed in the presence of sodium acetate (pH 3.5), EDTA, and phenylmethylsulfonylfhioride (PMSF) over about 30 minutes.
- Cell debris can be removed by centrifugation.
- supernatants from such expression systems are generally first concentrated using a commercially available protein concentration filter, for example, an Amicon or Millipore Pellicon ultrafiltration unit.
- a protease inhibitor such as PMSF may be included in any of the foregoing steps to inhibit proteolysis and antibiotics may be included to prevent the growth of adventitious
- the antibody or antibody fragment compositions prepared from such cells can be purified using, for example, hydroxylapatite chromatography, gel electrophoresis, dialysis, and affinity chromatography, with affinity chromatography being the preferred purification technique.
- affinity chromatography is the preferred purification technique.
- the suitability of protein A as an affinity ligand depends on the species and isotype of any immunoglobulin Fc domain that is present in the antibody.
- Protein A can be used to purify antibodies or antibody fragments that are based on human 1, 2, or 4 heavy chains (Lindmark et al., J. Immunol. Meth. 62: 1-13 (1983)).
- Protein G is recommended for all mouse isotypes and for human 3 heavy chain antibodies or antibody fragments (Guss et al., EMBO J.
- the matrix to which the affinity ligand is attached is most often agarose, but other matrices are available.
- Mechanically stable matrices such as controlled pore glass or poly(styrene-divinyl)benzene allow for faster flow rates and shorter processing times than can be achieved with agarose.
- the antibodies or antibody fragments comprise a C H 3 domain
- the Bakerbond ABXTMresin J. T. Baker, Phillipsburg, NJ
- the mixture comprising the antibody or antibody fragment of interest and contaminants may be subjected to low pH hydrophobic interaction chromatography using an elution buffer at a pH between about 2.5- 4.5, preferably performed at low salt concentrations ⁇ e.g., from about 0-0.25 M salt).
- Anti-HCV antibodies provided herein may be identified, screened for, or characterized for their physical/chemical properties and/or biological activities by various assays known in the art. Anti-HCV antibodies with certain biological characteristics may be selected as described in the Examples.
- an anti-HCV antibody of the invention is tested for its antigen binding activity, e.g., by known methods such as ELISA, Western blot, etc.
- competition assays may be used to identify an anti-HCV antibody that competes with any anti-HCV antibody described herein (e.g., V362) for binding to HCV E2 protein.
- competitive binding may be determined using an Enzyme-Linked Immunosorbent Assay (ELISA) assay.
- ELISA Enzyme-Linked Immunosorbent Assay
- an antibody that binds to HCV E2 protein competitively with an anti-HCV antibody comprising a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23, and a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25.
- an antibody is provided that binds to HCV E2 protein competitively with an anti-HCV antibody comprising the heavy chain amino acid sequence shown in SEQ ID NOs:27 or 28 and the light chain amino acid sequence shown in SEQ ID NO:30.
- such a competing antibody binds to the same epitope (e.g., a linear or a conformational epitope) that is bound by any anti-HCV antibody described herein (e.g., V362).
- epitope e.g., a linear or a conformational epitope
- V362 anti-HCV antibody described herein
- Detailed exemplary methods for mapping an epitope to which an antibody binds are provided in Morris (1996) "Epitope Mapping Protocols," in Methods in Molecular Biology vol. 66 (Humana Press, Totowa, NJ).
- immobilized HCV E2 protein is incubated in a solution comprising a first labeled antibody that binds to HCV E2 protein (e.g., V362) and a second unlabeled antibody that is being tested for its ability to compete with the first antibody for binding to HCV E2 protein.
- the second antibody may be present in a hybridoma supernatant.
- immobilized HCV E2 protein is incubated in a solution comprising the first labeled antibody but not the second unlabeled antibody. After incubation under conditions permissive for binding of the first antibody to HCV E2 protein, excess unbound antibody is removed, and the amount of label associated with immobilized HCV E2 protein is measured. If the amount of label associated with immobilized HCV E2 protein is
- assays are provided for identifying anti-HCV antibodies as described herein having biological activity.
- Biological activity may include, e.g., neutralization of HCV pseudoparticles (HCVpp), neutralization of infectious cell culture HCV (HCVcc), inhibition of HCV infection, etc.
- Antibodies having such biological activity in vivo and/or in vitro are also provided.
- an antibody of the invention is tested for such biological activity.
- Biological activity can be expressed as 90% effective concentration (EC90) values as described in Hotzel et al., Protein Eng Des Sel. (2011), 24:679-89 and Diao et al., J Virol. (2012), 86(20): 10935-49.
- antibodies can also be screen for their ability to neutralize an HCV infection.
- neutralization of an HCV infection is based on a HCV pseudotyped particles (HCVpp) neutralization assay as described herein.
- HCVpp infection is determined using the Luciferase Assay System (Promega) which detects the luciferase expressed by HCVpp and allows measurement of inhibition of HCV infection and spread for calculation of the anti-HCV antibody EC90 value.
- neutralization of an HCV infection is based on a
- HCVcc recombinant cell culture-derived HCV neutralization assay infecting human hepatoma cell lines as described herein.
- neutralization of HCVcc expressing wild-type genotype 2a Jcl is conducted as previously described in Hotzel et al., Protein Eng Des Sel. (2011), 24:679-89 and Diao et al., J Virol. (2012), 86(20): 10935-49, to test the ability of the anti-HCV antibodies to inhibit entry and infection of HCV.
- Multiplicity of infection (MOI) for Jcl HCVcc is calculated based on virus titer as measured by TCID 50 calculations for obtainment of an EC90 value.
- an anti-HCV antibody provided herein has an EC90 of ⁇ 500 ⁇ g/mL, ⁇ 250 ⁇ g/mL, ⁇ 100 ⁇ g/mL, ⁇ 10 ⁇ g/mL, ⁇ 1 ⁇ g/mL, ⁇ 0.1 ⁇ g/mL, ⁇ 0.01 ⁇ g/mL, or ⁇ 0.001 ⁇ g/mL..
- an anti-HCV antibody provided herein has an EC90 of less than about any of about 100 ⁇ g/mL, 95 ⁇ g/mL, 90 ⁇ g/mL, 85 ⁇ g/mL, 80 ⁇ g/mL, 75 ⁇ g/mL, 70 ⁇ g/mL, 65 ⁇ g/mL, 60 ⁇ g/mL, 55 ⁇ g/mL, 50 ⁇ g/mL, 45 ⁇ g/mL, 40 ⁇ g/mL, 35 ⁇ g/mL, 30 ⁇ g/mL, 25 ⁇ g/mL, 20 ⁇ g/mL, 15 ⁇ g/mL, 10 ⁇ g/mL, 9 ⁇ g/mL, 8 ⁇ g/mL, 7 ⁇ g/mL, 6 ⁇ g/mL, 5 ⁇ g/mL, 4 ⁇ g/mL, 3 ⁇ g/mL, 2 ⁇ g/mL, 1 ⁇ g/mL, 0.9
- an anti-HCV antibody described herein has a lower EC90 (for example as measured in an HCVpp or HCVcc assay described herein) as compared to the EC90 of a parent antibody (such as MRCTlO. l) described herein for inhibition of HCV infection.
- the antibody has an EC90 of less than about 5 ⁇ g/ml, less than about 4 ⁇ g/ml, less than about 3 ⁇ g/ml, less than about 2 ⁇ g/ml, or about 1 ⁇ g/ml for inhibition of infection of an HCV genotype.
- the EC90 value of an anti-HCV antibody as described herein is less than any of 50%, 40%, 30%, 20%, 15%, 10%, 5%, or 1%, inclusive, including any value in between these numbers, of the EC90 value of MRCTlO. l for inhibition of infection by an HCV genotype selected from the group consisting of genotype 1 (e.g., genotype la and genotype lb), genotype 2 (e.g.
- the HCV genotype is an HCV quasispecies.
- the term "quasispecies" refers to a viral population that contains a different genetic sequence as a result of the incorporation or deletion of alternative or additional gene sequences, either correct or erroneous. Quasispecies may result, for example, from the genetic copy process of other gene sequences that are already present. Quasispecies may also arise in the context of the evolutionary process, in response to a host immune response, or in response to a therapeutic treatment. As used herein, quasispecies include both the variant species and the wild-type species from which the variant species derived. D. Methods and Compositions for Diagnostics and Detection
- any of the anti-HCV antibodies provided herein is useful for detecting the presence of HCV E2 protein or fragment thereof in a biological sample.
- the term "detecting" as used herein encompasses quantitative or qualitative detection.
- a biological sample comprises blood, serum, a cell or tissue, such as liver tissue from a liver biopsy.
- an anti-HCV antibody for use in a method of diagnosis or detection is provided.
- a method of detecting the presence of HCV in a biological sample comprises contacting the biological sample with an anti-HCV antibody as described herein under conditions permissive for binding of the anti-HCV antibody to HCV, and detecting whether a complex is formed between the anti-HCV antibody and HCV.
- Such method may be an in vitro or in vivo method.
- a method of detecting the presence of HCV E2 protein or fragment thereof in a biological sample is provided.
- the method comprises contacting the biological sample with an anti-HCV antibody as described herein under conditions permissive for binding of the anti-HCV antibody to HCV E2 protein, and detecting whether a complex is formed between the anti-HCV antibody and HCV E2 protein.
- an anti-HCV antibody is used to select subjects eligible for therapy with an anti-HCV antibody, e.g. where HCV or HCV E2 protein is a biomarker for selection of patients.
- a diagnostic test apparatus and method for determining or detecting the presence of HCV in a sample may comprise, as a reagent, one or more anti-HCV antibodies or fragments thereof as described herein.
- the antibody/ies may, for example, be immobilized on a solid support (e.g., on a microtiter assay plate, or on a particulate support) and serve to "capture" HCV particles from a sample (e.g., a blood or serum sample or other clinical specimen - such as a liver biopsy).
- the captured virus particles may then be detected by, for example, adding a further, labeled, reagent which binds to the captured virus particles.
- the assay may take the form of an ELISA, especially a sandwich-type ELISA, but any other assay format could in principle be adopted (e.g., radioimmunoassay, Western blot) including immunochromatographic or dipstick-type assays.
- any other assay format could in principle be adopted (e.g., radioimmunoassay, Western blot) including immunochromatographic or dipstick-type assays.
- the anti-HCV antibodies or fragments thereof as described herein may either be labeled or unlabelled.
- Unlabelled antibodies can be used in combination with other labeled antibodies (second antibodies).
- the antibodies can be directly labeled.
- a wide variety of labels may be employed - such as radionuclides, fluors, enzymes, enzyme substrates, enzyme cofactors, enzyme inhibitors, ligands (particularly haptens), etc.
- Labels include, but are not limited to, labels or moieties that are detected directly (such as fluorescent, chromophoric, electron-dense, chemiluminescent, and radioactive labels), as well as moieties, such as enzymes or ligands, that are detected indirectly, e.g., through an enzymatic reaction or molecular interaction.
- Exemplary labels include, but are not limited to, the radioisotopes 32 P, 14 C, 125 I, 3 H, and 131 I, fluorophores such as rare earth chelates or fluorescein and its derivatives, rhodamine and its derivatives, dansyl, umbelliferone, luceriferases, e.g., firefly luciferase and bacterial luciferase (U.S. Patent No.
- luciferin 2,3-dihydrophthalazinediones
- horseradish peroxidase HRP
- alkaline phosphatase alkaline phosphatase
- ⁇ -galactosidase glucoamylase
- lysozyme saccharide oxidases, e.g., glucose oxidase, galactose oxidase, and glucose-6-phosphate dehydrogenase
- heterocyclic oxidases such as uricase and xanthine oxidase, coupled with an enzyme that employs hydrogen peroxide to oxidize a dye precursor such as HRP, lactoperoxidase, or microperoxidase, biotin/avidin, spin labels, bacteriophage labels, stable free radicals, and the like.
- the assay apparatus and corresponding method should be capable of detecting in a sample HCV representative from any of these genotypes or quasispecies of these genotypes.
- the sample is compared to a control sample.
- the control sample is from an individual known to be infected with HCV.
- the individual is known to infected with one or more HCV genotypes selected from the group consisting of genotype 1 ⁇ e.g., genotype la and genotype lb), genotype 2 ⁇ e.g., genotype 2a, genotype 2b, genotype 2c), genotype 3 ⁇ e.g., genotype 3a), genotype 4 ⁇ e.g., genotype 4a), genotype 5, and genotype 6.
- the individual is known to be infected with one or more HCV quasispecies. In some embodiments, the individual is known to be infected with one or more HCV quasispecies. In some embodiments, genotype 1 ⁇ e.g., genotype la and genotype lb), genotype 2 ⁇ e.g., genotype 2a, genotype 2b, genotype 2c), genotype 3 ⁇ e.g., genotype 3a), genotype 4 ⁇ e
- control sample is from an individual known not to be infected with HCV.
- any of the methods of treatment described are based on the determination or detection of HCV in a sample by any of the anti-HCV antibodies or fragments thereof described herein.
- "based upon” includes (1) assessing, determining, or measuring the subject's characteristics as described herein (and preferably selecting a subject suitable for receiving treatment); and (2) administering the treatment(s) as described herein.
- a method is provided for identifying an individual suitable or not suitable (unsuitable) for treatment with the anti-HCV antibody or fragment thereof.
- an individual suitable for treatment is administered an anti-HCV antibody as disclosed herein.
- a method is providing for selecting or not selecting an individual for treatment with the anti-HCV antibody or fragment thereof, the method comprising: a) assessing the viral load and/or viral titer in a biological sample from the individual, and b) selecting the individual for treatment with an anti-HCV antibody or fragment thereof if the viral load is at least 5 IU/mL.
- the viral load is at least 5 IU/mL, 10 IU/mL, 20 IU/mL, 40 IU/mL, 80 IU/mL, 100 IU/mL, 500 IU/mL, 1,000 IU/mL, 10,000 IU/mL, 100,000 IU/mL, 200,000 IU/mL, 300,000 IU/mL, 400,000 IU/mL, 500,000 IU/mL, 600,000 IU/mL, 700,000 IU/mL, 800,000 IU/mL, 900,000 IU/mL, or 1,000,000 IU/mL, inclusive, including any values in between these numbers.
- Exemplary disorders that may be diagnosed using an antibody of the invention include acute HCV infection and chronic HCV infection.
- an assay method for identifying an agent that improves or enhances the efficacy of the neutralizing activity of the anti-HCV antibody or fragment thereof against hepatitis C virus comprising the steps of: (a) contacting said anti-HCV antibody or antigen binding fragment thereof with an agent to be tested; and (b) determining whether the agent improves or enhances the efficacy of the anti-HCV antibody or antigen binding fragment thereof in neutralizing the infectivity of hepatitis C virus.
- the ability of the agent to improve or enhance the efficacy of the neutralizing activity of the anti-HCV antibody or fragment thereof against hepatitis C virus is compared to a control.
- the control is the anti-HCV antibody or fragment thereof in the absence of the agent.
- the control is humanized antibody or fragment thereof with a placebo, e.g., water, saline, sugar water, etc.
- agent may be a single entity or it may be a combination of entities.
- the agent may be an organic compound or other chemical.
- the agent may be a compound, which is obtainable from or produced by any suitable source, whether natural or artificial.
- the agent may be an amino acid molecule, a polypeptide, or a chemical derivative thereof, or a combination thereof.
- the agent may even be a polynucleotide molecule - which may be a sense or an anti-sense molecule.
- the agent is an antibody.
- the agent is a cytokine (such as interferon- ).
- the agent is a direct acting antiviral agent.
- the direct acting antiviral agent is viral protease inhibitor or a viral polymerase inhibitor.
- the agent is an indirect acting viral agent.
- the agent may be designed or obtained from a library of compounds, which may comprise peptides, as well as other compounds, such as small organic molecules.
- the agent may be a natural substance, a biological
- a macromolecule or an extract made from biological materials such as bacteria, fungi, or animal (particularly mammalian) cells or tissues, an organic or an inorganic molecule, a synthetic agent, a semi- synthetic agent, a structural or functional mimetic, a peptide, a peptidomimetics, a derivatized agent, a peptide cleaved from a whole protein, or a peptides synthesized synthetically (such as, by way of example, either using a peptide synthesizer or by recombinant techniques or combinations thereof, a recombinant agent, an antibody, a natural or a non-natural agent, a fusion protein or equivalent thereof and mutants, derivatives or combinations thereof.
- the agent will be an organic compound.
- the organic compounds will comprise two or more hydrocarbyl groups.
- hydrocarbyl group means a group comprising at least C and H and may optionally comprise one or more other suitable substituents. Examples of such substituents may include halo-, alkoxy-, nitro-, an alkyl group, a cyclic group etc.
- substituents may include halo-, alkoxy-, nitro-, an alkyl group, a cyclic group etc.
- a combination of substituents may form a cyclic group. If the hydrocarbyl group comprises more than one C then those carbons need not necessarily be linked to each other. For example, at least two of the carbons may be linked via a suitable element or group.
- the hydrocarbyl group may contain hetero atoms. Suitable hetero atoms will be apparent to those skilled in the art and include, for instance, sulphur, nitrogen and oxygen.
- the agent comprises at least one cyclic group.
- the cyclic group may be a polycyclic group, such as a non-fused polycyclic group.
- the agent comprises at least the one of said cyclic groups linked to another hydrocarbyl group.
- the agent may contain halo groups.
- halo means fluoro, chloro, bromo or iodo.
- the agent may contain one or more of alkyl, alkoxy, alkenyl, alkylene and alkenylene groups - which may be unbranched- or branched-chain.
- compositions and formulations of an anti-HCV antibody as described herein are prepared by mixing such antibody having the desired degree of purity with one or more optional pharmaceutically acceptable carriers ⁇ Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980)), in the form of lyophilized formulations or aqueous solutions.
- Pharmaceutically acceptable carriers are generally nontoxic to recipients at the dosages and concentrations employed, and include, but are not limited to: buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride;
- hexamethonium chloride benzalkonium chloride; benzethonium chloride; phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol;
- polypeptides such as serum albumin, gelatin, or immunoglobulins
- proteins such as serum albumin, gelatin, or immunoglobulins
- hydrophilic polymers such as polyvinylpyrrolidone
- amino acids such as glycine, glutamine, asparagine, histidine, arginine, or lysine
- monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins chelating agents such as EDTA; sugars such as sucrose, mannitol, trehalose or sorbitol; salt-forming counter-ions such as sodium; metal complexes ⁇ e.g.
- sHASEGP soluble neutral-active hyaluronidase glycoproteins
- rHuPH20 HYLENEX ® , Baxter International, Inc.
- a sHASEGP is combined with one or more additional glycosaminoglycanases such as chondroitinases.
- Buffers are used to control the pH in a range which optimizes the therapeutic effectiveness, especially if stability is pH dependent. Buffers are preferably present at concentrations ranging from about 50 mM to about 250 mM. Suitable buffering agents for use with the present invention include both organic and inorganic acids and salts thereof, such as citrate, phosphate, succinate, tartrate, fumarate, gluconate, oxalate, lactate, acetate. Additionally, buffers may comprise histidine and trimethylamine salts such as Tris.
- Preservatives are added to retard microbial growth, and are typically present in a range from 0.2% - 1.0% (w/v). Suitable preservatives for use with the present invention include octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride;
- benzalkonium halides e.g. , chloride, bromide, iodide), benzethonium chloride; thimerosal, phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol; cyclohexanol, 3-pentanol, and m-cresol.
- Tonicity agents sometimes known as “stabilizers” are present to adjust or maintain the tonicity of liquid in a composition. When used with large, charged biomolecules such as proteins and antibodies, they are often termed “stabilizers” because they can interact with the charged groups of the amino acid side chains, thereby lessening the potential for inter- and intra- molecular interactions. Tonicity agents can be present in any amount between 0.1% to 25% by weight, or more preferably between 1% to 5% by weight, taking into account the relative amounts of the other ingredients.
- Preferred tonicity agents include polyhydric sugar alcohols, preferably trihydric or higher sugar alcohols, such as glycerin, erythritol, arabitol, xylitol, sorbitol and mannitol.
- Non-ionic surfactants or detergents are present to help solubilize the therapeutic agent as well as to protect the therapeutic protein against agitation-induced aggregation, which also permits the formulation to be exposed to shear surface stress without causing denaturation of the active therapeutic protein or antibody.
- Non-ionic surfactants are present in a range of about 0.05 mg/ml to about 1.0 mg/ml, preferably about 0.07 mg/ml to about 0.2 mg/ml.
- Suitable non-ionic surfactants include polysorbates (20, 40, 60, 65, 80, etc.), polyoxamers (184, 188, etc.), PLURONIC ® polyols, TRITON ® , polyoxyethylene sorbitan monoethers (TWEEN ® -20, TWEEN ® -80, etc.), lauromacrogol 400, polyoxyl 40 stearate, polyoxyethylene hydrogenated castor oil 10, 50 and 60, glycerol monostearate, sucrose fatty acid ester, methyl cellulose and carboxymethyl cellulose.
- Anionic detergents that can be used include sodium lauryl sulfate, dioctyle sodium sulfo succinate and dioctyl sodium sulfonate.
- Cationic detergents include benzalkonium chloride or benzethonium chloride.
- compositions may comprise as - or in addition to - the carrier, excipient or dilutent any suitable binder(s), lubricant(s), suspending agent(s), coating agent(s) or solubilizing agent(s).
- compositions useful in the present invention may be formulated to be administered using a mini-pump or by a mucosal route, for example, as a nasal spray or aerosol for inhalation or ingestible solution, or parenterally in which the composition is formulated by an injectable form, for delivery, by, for example, an intravenous, intramuscular or subcutaneous route.
- the formulation may be designed to be administered by a number of routes.
- an anti-HCV antibody formulation is a lyophilized anti-HCV antibody formulation.
- an anti-HCV antibody formulation is an aqueous anti-HCV antibody formulation.
- Exemplary lyophilized antibody formulations are described in US Patent No. 6,267,958.
- Aqueous antibody formulations include those described in US Patent No. 6,171,586 and WO2006/044908, the latter formulations including a histidine-acetate buffer.
- the formulation herein may also contain more than one active ingredients as necessary for the particular indication being treated, preferably those with complementary activities that do not adversely affect each other.
- an active ingredient is a therapeutic agent.
- a therapeutic agent such as a viral protease inhibitor, a viral polymerase inhibitor, a NS5A inhibitor, an interferon, a cyclophilin inhibitor, or an antibody.
- Viral protease inhibitors include NS3-4a protease inhibitors such as, but not limited to, Teleprevir, Boceprevir, Semeprevir (TMC435), BI201335, Danoprevir/r (RG7227), Vaniprevir (MK-7009), Narlaprevir (SCH 900518), Asunaprevir, GS-9256, GS-9451, ABT-450/r, ACH-1625, ACH-2684, and MK-5172.
- Viral polymerase inhibitors include nucleoside/nucleotide analogue inhibitors and non-nucleoside inhibitors of HCV RNA-dependent RNA polymerase. Such viral polymerase inhibitors include, but are not limited to, PSI-7977, Mercitabine, IDX184, PSI-938, INX-189,
- HCV NS5A inhibitors include, but are not limited to, Daclatasvir, PPI-461, GS-5885, and
- cyclophilin inhibitors contemplated herein include, but are not limited to, Alisporivir and SCY-465.
- An interferon contemplated herein include, but are not limited to, interferon-a (such as interferon-a 2a or interferon-a 2b) and interferon-a derivatives (such as pegylated interferon- ⁇ 2a or pegylated interferon- ⁇ 2b).
- Antibodies that bind to other HCV proteins required by HCV to infect the cell are also contemplated.
- the therapeutic agent is ribavirin.
- a therapeutic agent as described herein can be used in a formulation with an anti-HCV antibody as described herein including anti-HCV antibodies described in Owsianka et al., J. Gen.
- Such active ingredients are suitably present in combination in amounts that are effective for the purpose intended.
- Active ingredients may be entrapped in microcapsules prepared, for example, by coacervation techniques or by interfacial polymerization, for example,
- microcapsules respectively, in colloidal drug delivery systems (for example, liposomes, albumin microspheres, microemulsions, nano-particles and nanocapsules) or in
- Stability of the proteins and antibodies described herein may be enhanced through the use of non-toxic "water-soluble polyvalent metal salts".
- water-soluble polyvalent metal salts examples include Ca 2+ , Mg 2+ , Zn 2+ , Fe 2+ , Fe 3+ , Cu 2+ , Sn 2+ , Sn 4+ , Al 2+ and Al 3+ .
- Exemplary anions that can form water soluble salts with the above polyvalent metal cations include those formed from inorganic acids and/or organic acids.
- Such water-soluble salts have are soluble in water (at 20°C) to at least about 20 mg/ml, alternatively at least about 100 mg/ml, alternatively at least about 200 mg/ml.
- Suitable inorganic acids that can be used to form the "water soluble polyvalent metal salts" include hydrochloric, acetic, sulfuric, nitric, thiocyanic and phosphoric acid.
- Suitable organic acids that can be used include aliphatic carboxylic acid and aromatic acids. Aliphatic acids within this definition may be defined as saturated or unsaturated C 2 -9 carboxylic acids ⁇ e.g., aliphatic mono-, di- and tri-carboxylic acids).
- exemplary monocarboxylic acids within this definition include the saturated C 2 -9 monocarboxylic acids acetic, proprionic, butyric, valeric, caproic, enanthic, caprylic pelargonic and capryonic, and the unsaturated C 2 -9 monocarboxylic acids acrylic, propriolic methacrylic, crotonic and isocro tonic acids.
- exemplary dicarboxylic acids include the saturated C 2 -9 dicarboxylic acids malonic, succinic, glutaric, adipic and pimelic, while unsaturated C 2 -9 dicarboxylic acids include maleic, fumaric, citraconic and mesaconic acids.
- Exemplary tricarboxylic acids include the saturated C 2 -9 tricarboxylic acids tricarballylic and 1,2,3-butanetricarboxylic acid. Additionally, the carboxylic acids of this definition may also contain one or two hydroxyl groups to form hydroxy carboxylic acids. Exemplary hydroxy carboxylic acids include glycolic, lactic, glyceric, tartronic, malic, tartaric and citric acid. Aromatic acids within this definition include benzoic and salicylic acid.
- Commonly employed water soluble polyvalent metal salts which may be used to help stabilize the encapsulated polypeptides of this invention include, for example: (1) the inorganic acid metal salts of halides (e.g., zinc chloride, calcium chloride), sulfates, nitrates, phosphates and thiocyanates; (2) the aliphatic carboxylic acid metal salts (e.g., calcium acetate, zinc acetate, calcium proprionate, zinc glycolate, calcium lactate, zinc lactate and zinc tartrate); and (3) the aromatic carboxylic acid metal salts of benzoates (e.g., zinc benzoate) and salicylates.
- halides e.g., zinc chloride, calcium chloride
- sulfates e.g., nitrates, phosphates and thiocyanates
- aliphatic carboxylic acid metal salts e.g., calcium acetate, zinc acetate, calcium proprionate, zinc glycolate, calcium lac
- compositions of anti-HCV antibodies can be designed to
- sustained-release formulations immediately release an anti-HCV antibody ("immediate-release” formulations), to gradually release the antibodies over an extended period of time (“sustained-release,” “controlled- release,” or “extended-release” formulations), or with alternative release profiles.
- the additional materials used to prepare a pharmaceutical formulation can vary depending on the therapeutic form of the formulation (e.g., whether the system is designed for immediate- release or sustained-, controlled-, or extended-release).
- a sustained- release formulation can further comprise an immediate-release component to quickly deliver a priming dose following drug delivery, as well as a sustained-release component.
- sustained-release formulations can be combined with immediate-release formulations to provide a rapid "burst" of drug into the system as well as a longer, gradual release.
- a core sustained-release formulation may be coated with a highly soluble layer incorporating the drug.
- a sustained-release formulation and an immediate- release formulation may be included as alternate layers in a tablet or as separate granule types in a capsule.
- Other combinations of different types of drug formulations can be used to achieve the desired therapeutic plasma profile.
- sustained-release preparations include semi-permeable matrices of solid hydrophobic polymers containing the antagonist, which matrices are in the form of shaped articles, e.g., films, or microcapsules.
- sustained-release matrices include polyesters, hydrogels (for example, poly(2-hydroxyethyl-methacrylate), or poly(vinylalcohol)), polylactides (U.S. Pat. No.
- copolymers of L-glutamic acid and ethyl-L-glutamate non-degradable ethylene-vinyl acetate
- degradable lactic acid-glycolic acid copolymers such as the LUPRON DEPOTTM (injectable microspheres composed of lactic acid-glycolic acid copolymer and leuprolide acetate), and poly-D-(-)-3-hydroxybutyric acid.
- the formulations to be used for in vivo administration are generally sterile. Sterility may be readily accomplished, e.g., by filtration through sterile filtration membranes.
- the pharmaceutical compositions may be used in any of the methods described herein.
- the pharmaceutical composition may be used among those subjects (e.g., humans) susceptible to infection with HCV i.e. to prevent or reduce/decrease the onset of HCV infection.
- the pharmaceutical composition may be used among those subjects (e.g., humans) already infected with HCV i.e. to treat HCV infection. Such treatment may facilitate clearance of the virus from those subjects who are acutely or chronically infected including infected patients undergoing liver transplantation.
- the pharmaceutical composition is used to prevent liver transplant re-infection.
- the pharmaceutical composition is used to prevent HCV infection of the transplanted liver.
- the pharmaceutical composition is used to prevent infection of a patient receiving an HCV infected liver transplant.
- the pharmaceutical composition is administered by an individual before receiving a liver transplant.
- the pharmaceutical composition is administered by an individual during receipt of a liver transplant.
- the pharmaceutical composition is administered by an individual after receiving a liver transplant.
- the invention provides a method for the treatment and/or prevention of hepatitis C virus infection, comprising the use of the anti-HCV antibody or the anti-HCV antibody fragment or the pharmaceutical composition.
- an effective amount of the anti-HCV antibody or antibody fragment thereof or the pharmaceutical composition is administered to the subject.
- the anti-HCV antibody or anti-HCV antibody fragment is administered in a therapeutic effective amount to effect beneficial clinical results, including, but not limited to ameliorating one or more symptoms of HCV infections or aspects of HCV infection.
- the anti-HCV antibody or anti-HCV antibody fragment is administered in a therapeutic effective amount to reduce viral titer and/or viral load of HCV.
- the anti-HCV antibody or anti- HCV antibody fragment is administered in a therapeutic effective amount to achieve a sustained virologic response.
- sustained virologic response refers to the absence of detectable viremia twelve weeks after stopping anti-HCV treatment.
- an anti-HCV antibody or a fragment thereof or the pharmaceutical composition for use in the treatment and/or prevention of hepatitis C virus infection in a subject.
- an antibody of a fragment thereof or the pharmaceutical composition in the manufacture of a composition for the treatment and/or prevention of hepatitis C virus infection in a subject.
- the antibody/ies may be administered, for example, in the form of immune serum or may more preferably be a purified recombinant or monoclonal antibody. Methods of producing sera or monoclonal antibodies with the desired specificity are routine and well- known to those skilled in the art.
- the antibody/ies can be administered by various routes including, for example, injection, intubation, via a suppository, orally or topically, the latter of which can be passive, for example, by direct application of an ointment or powder containing the antibodies, or active, for example, using a nasal spray or inhalant.
- the antibodies can also be administered as a topical spray, if desirable, in which case one component of the composition is an appropriate propellant.
- anti-HCV antibodies and fragments thereof described herein can be any anti-HCV antibodies and fragments thereof described herein.
- administration e.g., as a bolus or by continuous infusion over a period of time, by
- subcutaneous, intramuscular, intraperitoneal, intracerobrospinal, intra-articular, intrasynovial, intrathecal, or inhalation routes generally by intravenous or subcutaneous administration.
- a passive immunization regime may conveniently comprise administration of the anti-HCV antibody of fragment thereof as described herein and/or administration of antibody in combination with other antiviral therapeutic compounds.
- passive immunization techniques have been used safely to treat HIV infection (Armbruster et al, J. Antimicrob. Chemother. 54, 915-920 (2004); Stiegler & Katinger, J. Antimicrob. Chemother. 51, 757-759 (2003)).
- the active or passive immunization methods of the invention should allow for the protection or treatment of individuals against infection with viruses of any of genotypes 1-6 of HCV.
- the anti-HCV antibody or fragment thereof can be administered in combination with a second therapeutic agent (such as a viral protease inhibitor).
- the second therapeutic agent is an antiviral therapeutic agent.
- the anti-HCV antibody or fragment thereof is administered in combination with, sequential to, concurrently with, consecutively with, rotationally with, or intermittently with a second therapeutic agent (such as a viral protease inhibitor).
- the administration of the combination of an anti-HCV antibody or fragment thereof and a second therapeutic agent ameliorates one or more symptom of HCV, reduces and/or suppresses viral titer and/or viral load, and/or prevents HCV, and/or achieves a sustained virologic response more than treatment with the anti-HCV antibody or fragment thereof or second therapeutic agent alone.
- a second therapeutic agent such as a viral protease inhibitor
- the anti-HCV antibody or fragment thereof and the second therapeutic agent are provided in separate dosage forms.
- the anti-HCV antibody or fragment thereof and the second therapeutic agent are provided in the same dosage form.
- the invention provides methods for treating or preventing an HCV infection in a subject comprising administering to the subject an effective amount of an anti-HCV antibody described herein.
- the subject is human.
- the subject has been diagnosed with HCV infection. Any of the anti-HCV antibodies provided herein may be used in therapeutic methods.
- the anti-HCV antibodies and fragments thereof or a pharmaceutical composition comprising same are useful in reducing, eliminating, or inhibiting HCV infection and can be used for treating any pathological condition that is characterized, at least in part, by HCV infection.
- the anti-HCV antibodies and fragments thereof and/or the pharmaceutical composition can be used for treating an HCV infection.
- the anti-HCV antibodies and fragments thereof and/or the pharmaceutical composition can also be used in methods for preventing a HCV infection.
- hepatitis C virus or "HCV” is well understood in the art and refers to a virus which is a member of the genus Hepacivirus of the family flaviviridae.
- HCV is a lipid enveloped virus having a diameter of approximately 55-65 nm in diameter with a positive strand RNA genome.
- the hepatitis C virus species is classified into six genotypes (1-6) with several subtypes within each genotype.
- the subject is infected with one or more HCV genotypes selected from the group consisting of genotype 1 (e.g., genotype la and genotype lb), genotype 2 (e.g., genotype 2a, genotype 2b, genotype 2c), genotype 3 (e.g., genotype 3a), genotype 4 (e.g., genotype 4a), genotype 5, and genotype 6.
- genotype 1 e.g., genotype la and genotype lb
- genotype 2 e.g., genotype 2a, genotype 2b, genotype 2c
- genotype 3 e.g., genotype 3a
- genotype 4 e.g., genotype 4a
- genotype 5 genotype 5
- genotype 6 is found in Southeast Asia and Australia.
- an anti-HCV antibody or fragment thereof as described herein are useful in methods of treating an individual infected with an HCV genotype attributed to a specific geographical population (such as genotype 2 in Japan). See Simmonds P., J Gen Virol., (2004)., 85(Pt 11):3173-88 for non-limiting examples of specific geographical populations that can be treated with the anti-HCV antibodies described herein. [0240] Provided herein are methods for treating a hepatitis C virus infection in a subject, comprising administering an effective amount of the anti-HCV antibody fragment thereof described herein. In some embodiments, the anti-HCV antibodies and fragments thereof described herein are useful in methods of treating an acute HCV infection.
- treating an acute HCV infection includes reducing, eliminating, or inhibiting an acute HCV infection.
- acute hepatitis C virus infection or "acute HCV infection,” as used herein, refers to the first 6 months after infection with HCV.
- a subject with an acute HCV infection will not develop any symptoms (i.e., free of acute HCV infection symptoms). Between 60% to 70% of subjects with acute HCV infection develop no symptoms during the acute phase. In some embodiments, a subject with acute HCV infection will develop symptoms.
- the methods of treatment described herein ameliorate (e.g. , reduce incidence of, reduce duration of, reduce or lessen severity of) of one or more symptoms of acute HCV infection.
- Symptoms of acute hepatitis C infection include decreased appetite, fatigue, abdominal pain, jaundice, itching, and flu-like symptoms.
- the subject with acute HCV infection is infected with HCV of the genotype 1. Treatment during the acute HCV injection of genotype 1 has a greater than 90% success rate with half the treatment time required for chronic infections.
- the anti-HCV antibodies and fragments thereof described are useful in methods of treating a chronic HCV infection.
- treating a chronic HCV infection includes reducing, eliminating, or inhibiting a chronic HCV infection.
- chronic hepatitis C virus infection or "chronic HCV infection,” as used herein, refers to an infection with HCV which persisting for more than six months.
- the methods of treatment described herein ameliorate (e.g. , reduce incidence of, reduce duration of, reduce or lessen severity of) of one or more symptoms of chronic HCV infection.
- Symptoms of chronic HCV infection include fatigue, marked weight loss, flu-like symptoms, muscle pain, joint pain, intermittent low-grade fevers, itching, sleep disturbances, abdominal pain (especially in the right upper quadrant), appetite changes, nausea, diarrhea, dyspepsia, cognitive changes, depression, headaches, and mood swings.
- signs and symptoms may appear that are generally caused by either decreased liver function or increased pressure in the liver circulation, a condition known as portal hypertension.
- liver cirrhosis Possible signs and symptoms of liver cirrhosis include ascites, bruising and bleeding tendency, bone pain, varices (especially in the stomach and esophagus), fatty stools (steatorrhea), jaundice, and a syndrome of cognitive impairment known as hepatic encephalopathy.
- the chronic HCV infection may result in hepatocellular carcinoma.
- Chronic HCV infection can be further divided into two types (either or both of which are included in the methods of treatment provided herein) chronic active HCV infection and chronic persistent HCV infection.
- Chronic active HCV infection is HCV which is cause active damage to the liver.
- Chronic persistent HCV infection is a chronic HCV infection which is not currently causing damage to the liver, although pre-existing liver damage may be present.
- HCV-associated diseases or “HCV-associated disorders” as used herein, refers to an infection with HCV or a disease or disorder that is associated with HCV infection such as liver disease. Accordingly, in some embodiments, an anti-HCV antibody or fragment as described herein prevents development of an HCV-associated disease. In some embodiments, an anti-HCV antibody or fragment as described herein prevents development of an HCV-associated disease. In some embodiments, an anti-HCV antibody or fragment as described herein prevents development of an HCV-associated disease.
- the HCV-associated disease is HCV infection.
- the HCV-associated disease is a liver disease such as, but not limited to, cirrhosis or
- an anti-HCV antibody or fragment as described herein is used in methods of treating an HCV-associated disease.
- the HCV-associated disease is HCV infection.
- the HCV-associated disease is a liver disease such as, but not limited to, cirrhosis or
- the anti-HCV antibodies or fragments thereof may be administered to the subject infected with HCV prior to, concurrent with, or subsequent to a liver transplant.
- an anti-HCV antibody or fragment thereof prevents liver transplant re-infection.
- an anti-HCV antibody or fragment thereof prevents HCV infection of the transplanted liver.
- the anti-HCV antibodies and fragments thereof described herein are useful in methods of treatment including suppressing one or more aspects of a HCV infection.
- the HCV infection is a chronic HCV infection.
- the HCV infection is an acute HCV infection.
- the methods described herein suppress a HCV- associated laboratory finding (e.g., ALAT, AST, and GGTP levels in blood), viral replication, viral titer, viral load, or viremia.
- the methods described herein suppress or reduce viral titer.
- “Viral titer” is known in the art and indicates the amount of virus in a given biological sample.
- the methods described herein suppress or reduce viremia.
- “Viremia” is known in the art as the presence of virus in the bloodstream and/or viral titer in a blood or serum sample.
- the methods described herein suppress or reduce viral load.
- “Viral load” refers to the amount of hepatitis C virus in a person's blood.
- hepatitis C viral load test (known as a viral RNA test or HCV RNA test) are usually expressed as International Units/mL (IU/mL) or RNA copies/mL.
- a subject with a hepatitis C viral load of 1 million IU/mL or more is considered to have a high viral load.
- Amount of virus e.g., viral titer or viral load
- Amount of virus are indicated by various measurements, including, but not limited to amount of viral nucleic acid, the presence of viral particles, replicating units (RU), plaque forming units (PFU).
- Amount of virus such as high viral load, low viral load or undetectable viral load can be defined according to a clinical acceptable parameter established by those skilled in the art.
- an undetectable viral load is defined by the limit of the assay for detecting HCV.
- amount of virus is determined per unit fluid, such as milliliters.
- amount of virus is determined per weight unit, such as grams. Methods for determining amount of virus are known in the art and are also described herein.
- the methods described herein result in a sustained virologic response for at least 12 weeks after stopping the treatment.
- the methods described herein reduce serum alanine aminotransferase (ALT) levels.
- the subject treated with the anti-HCV antibodies and fragments thereof described herein is at risk for rapid HCV infection progression.
- Factors that have been reported to influence the rate of HCV disease progression include age (increasing age associated with more rapid progression), gender (males have more rapid disease progression than females), alcohol consumption (associated with an increased rate of disease progression), HIV co-infection (associated with a markedly increased rate of disease progression), and fatty liver (the presence of fat in liver cells has been associated with an increased rate of disease progression).
- the anti-HCV antibodies and fragments thereof described herein can also be used in methods for preventing a HCV infection.
- the anti-HCV antibodies and fragments thereof described herein are useful in methods of preventing an acute HCV infection.
- the anti-HCV antibodies and fragments thereof described herein are useful in methods of preventing a chronic HCV infection.
- the anti-HCV antibodies and fragments can be used in methods for preventing a HCV infection in a subject susceptible to infection with HCV.
- the anti-HCV antibodies and fragments thereof can also be used in methods for preventing a HCV infection in a subject exposed to or potentially exposed to HCV. "Exposure" to HCV denotes an encounter or potential encounter with HCV which could result in an HCV infection.
- an exposed subject is a subject that has been exposed to HCV by a route by which HCV can be transmitted.
- the subject has been exposed to or potentially exposed to blood of a subject with an HCV infection or blood from a subject which may or may not be infected with HCV (i.e. , HCV infection status of the blood exposure is unknown).
- HCV is often transmitted by blood-to-blood contact.
- the subject has been exposed to or potentially exposed to HCV by, but not limited to, use of blood products (e.g., a blood transfusion), "needle stick” accidents, sharing drug needles, snorting drugs, a sexual partner, iatrogenic medical or dental exposure, needles used in body piercings and tattoos, or a child whose mother has an HCV infection.
- blood products e.g., a blood transfusion
- needle stick accidents
- sharing drug needles e.g., snorting drugs, a sexual partner, iatrogenic medical or dental exposure
- needles used in body piercings and tattoos or a child whose mother has an HCV infection.
- the anti-HCV antibodies and fragments thereof described herein will be administered at the time or within any of about one day, one week, or one month of the exposure or potential exposure to HCV.
- the subject is a human or chimpanzee. In some embodiments, the subject is a human.
- provided herein is a method for treating or preventing a HCV infection in a subject comprising administering an anti-HCV antibody or fragment (such as an antibody described herein including V361 and V362, and any other antibody that binds HCV E2 protein) in combination with a second therapeutic agent.
- an anti-HCV antibody or fragment such as an antibody described herein including V361 and V362, and any other antibody that binds HCV E2 protein
- the second therapeutic agent is an antiviral therapeutic agent.
- the second therapeutic agent is selected from the group consisting of: a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a second anti-HCV antibody, and a combination thereof.
- the second therapeutic agent is a viral protease inhibitor selected from the group consisting of Teleprevir,
- the second therapeutic agent is a viral polymerase inhibitor selected from the group consisting of PSI-7977, Mercitabine, IDX184, PSI-938, INX- 189, Tegobuvir, Filibuvir, Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055.
- the second therapeutic agent is a HCV NS5A inhibitor selected from the group consisting of: Daclatasvir, PPI-461, GS-5885, and GSK2336805.
- the second therapeutic agent is a cyclophilin inhibitor selected from the group consisting of: Alisporivir and SCY-465.
- the second therapeutic agent is pegylated interferon-a 2a or pegylated interferon-a 2b.
- the second therapeutic agent is an antibody as described herein.
- the second therapeutic agent is ribavirin.
- the method comprises administering the anti-HCV antibody or fragment thereof in combination with, sequential to, concurrently with, consecutively with, rotationally with, or intermittently with a second therapeutic agent.
- the method comprising administering the combination of an anti-HCV antibody or fragment thereof and a second therapeutic agent ameliorates one or more symptom of HCV, reduces and/or suppresses viral titer and/or viral load, and/or prevents HCV, and/or achieves a sustained virologic response for at least twelve weeks after stopping treatment more than treatment with the ant-HCV antibody or fragment thereof or second therapeutic agent alone.
- the viral in the subject has been reduced to an undetectable level after treatment.
- the treatment is an interferon-free treatment.
- the subject is not responsive to interferon treatment.
- the method comprises
- the method comprises administering an anti-HCV antibody or fragment thereof and an interferon to a subject that is not responsive to interferon treatment alone.
- subjects that not responsive to interferon treatment include subjects that have detectable HCV during the course of treatment with interferon.
- Also provided herein is a method of preventing developing resistance to treatment, comprising administering an anti-HCV antibody or fragment (such as an antibody described herein including V361 and V362, and any other antibody that binds HCV E2 protein) and a second therapeutic agent.
- the second therapeutic agent is an antiviral therapeutic agent.
- the second therapeutic agent is selected from the group consisting of: a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a second anti-HCV antibody, and a combination thereof.
- the second therapeutic agent is a viral protease inhibitor selected from the group consisting of Teleprevir, Boceprevir, Semeprevir (TMC435), BI201335, Danoprevir/r (RG7227), Vaniprevir (MK-7009), Narlaprevir (SCH 900518), Asunaprevir, GS-9256, GS- 9451, ABT-450/r, ACH-1625, ACH-2684, and MK-5172.
- the second therapeutic agent is a viral polymerase inhibitor selected from the group consisting of PSI- 7977, Mercitabine, IDX184, PSI-938, INX-189, Tegobuvir, Filibuvir, Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055.
- the second therapeutic agent is a HCV NS5A inhibitor selected from the group consisting of: Daclatasvir, PPI-461, GS- 5885, and GSK2336805.
- the second therapeutic agent is a cyclophilin inhibitor selected from the group consisting of: Alisporivir and SCY-465.
- the second therapeutic agent is pegylated interferon-a 2a or pegylated interferon-a 2b.
- the second therapeutic agent is an antibody as described herein.
- the second therapeutic agent is ribavirin.
- the method comprises administering the anti-HCV antibody or fragment thereof in combination with, sequential to, concurrently with, consecutively with, rotationally with, or intermittently with a second therapeutic agent.
- the viral resistance in the subject is undetectable or low. Methods of determining undetectable or low viral resistance are known in the art.
- HCV RNA extracted from a biological sample collected every day up to 12 weeks post treatment from an individual administered an anti-HCV antibody a described herein is sequenced and compared to a control sequence (such as a wild-type sequence) to determine if mutations have arisen in the HCV that is infecting the individual. No mutations or low copy numbers of mutations as compared to the whole sample are indicative that viral resistance in the subject receiving treatment with an anti-HCV antibody is undetectable or low. In some embodiments, the viral resistance in a subject receiving an anti-HCV antibody and a second therapeutic agent is undetectable or low.
- the viral resistance in a subject receiving an anti-HCV antibody and a second therapeutic agent is undetectable or low as compared to a subject receiving a second therapeutic agent alone. In some embodiments, the viral resistance in a subject receiving an anti-HCV antibody and a second therapeutic agent is undetectable or low as compared to a subject receiving an anti-HCV antibody alone.
- the invention provides for the use of an anti-HCV antibody in the manufacture or preparation of a medicament.
- the medicament is for treatment of HCV infection.
- a medicament comprising an anti- HCV antibody for use in a method of treating HCV infection comprises administering to an individual having an HCV infection an effective amount of the medicament comprising an anti-HCV antibody.
- the method further comprises administering to the individual an effective amount of at least one additional therapeutic agent.
- the invention provides for the use of an anti-HCV antibody as described herein (such as MRCT10.1 or V362) in combination with a second therapeutic agent in the manufacture or preparation of a medicament.
- the second therapeutic agent is an antiviral therapeutic agent.
- the second therapeutic agent is selected from the group consisting of: a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a second anti-HCV antibody, and a combination thereof.
- the second therapeutic agent is a viral protease inhibitor selected from the group consisting of Teleprevir, Boceprevir, Semeprevir (TMC435), BI201335,
- Danoprevir/r (RG7227), Vaniprevir (MK-7009), Narlaprevir (SCH 900518), Asunaprevir, GS-9256, GS-9451, ABT-450/r, ACH-1625, ACH-2684, and MK-5172.
- the second therapeutic agent is a viral polymerase inhibitor selected from the group consisting of PSI-7977, Mercitabine, IDX184, PSI-938, INX-189, Tegobuvir,
- Filibuvir Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055.
- the second therapeutic agent is a HCV NS5A inhibitor selected from the group consisting of: Daclatasvir, PPI-461, GS-5885, and GSK2336805.
- the second therapeutic agent is a cyclophilin inhibitor selected from the group consisting of: Alisporivir and SCY-465.
- the second therapeutic agent is pegylated interferon-a 2a or pegylated interferon-a 2b.
- the second therapeutic agent is an antibody as described herein.
- the second therapeutic agent is ribavirin.
- Such combination therapies noted above encompass combined administration (where two or more therapeutic agents are included in the same or separate formulations), and separate administration, in which case, administration of the antibody of the invention can occur prior to, simultaneously, and/or following, administration of the additional therapeutic agent and/or adjuvant.
- the invention provides pharmaceutical formulations as described herein comprising any of the anti-HCV antibodies provided herein, e.g., for use in any of the above therapeutic methods.
- An anti-HCV antibody of the invention can be administered by any suitable means, including parenteral, intrapulmonary, and intranasal, and, if desired for local treatment, intralesional administration.
- Parenteral infusions include intramuscular, intravenous, intraarterial, intraperitoneal, or subcutaneous administration. Dosing can be by any suitable route, e.g. by injections, such as intravenous or subcutaneous injections, depending in part on whether the administration is brief or chronic.
- Various dosing schedules including but not limited to single or multiple administrations over various time- points, bolus administration, and pulse infusion are contemplated herein.
- Anti-HCV antibodies of the invention would be formulated, dosed, and administered in a fashion consistent with good medical practice. Factors for consideration in this context include the particular disorder being treated, the particular mammal being treated, the clinical condition of the individual patient, the cause of the disorder, the site of delivery of the agent, the method of administration, the scheduling of administration, and other factors known to medical practitioners.
- the antibody need not be, but is optionally formulated with one or more agents currently used to prevent or treat the disorder in question. The effective amount of such other agents depends on the amount of antibody present in the formulation, the type of disorder or treatment, and other factors discussed above. These are generally used in the same dosages and with administration routes as described herein, or about from 1 to 99% of the dosages described herein, or in any dosage and by any route that is
- an antibody of the invention when used alone or in combination with one or more other additional therapeutic agents, will depend on the type of disease to be treated, the type of antibody, the severity and course of the disease, whether the antibody is administered for preventive or therapeutic purposes, previous therapy, the patient's clinical history and response to the antibody, and the discretion of the attending physician.
- the antibody is suitably administered to the patient at one time or over a series of treatments. Depending on the type and severity of the disease, about 1 ⁇ g/kg to 15 mg/kg (e.g. O.
- lmg/kg-lOmg/kg can be an initial candidate dosage for administration to the patient, whether, for example, by one or more separate administrations, or by continuous infusion.
- One typical daily dosage might range from about 1 ⁇ g/kg to 100 mg/kg or more, depending on the factors mentioned above.
- the treatment would generally be sustained until a desired suppression of disease symptoms occurs.
- One exemplary dosage of the antibody would be in the range from about 0.05 mg/kg to about 10 mg/kg.
- one or more doses of about 0.5 mg/kg, 2.0 mg/kg, 4.0 mg/kg or 10 mg/kg (or any combination thereof) may be administered to the patient.
- Such doses may be administered intermittently, e.g.
- An exemplary dosing regimen comprises administering a monthly subcutaneous dose of 1 to 2 mg/kg anti-HCV antibody to an individual for up to six months.
- the individual has chronic HCV.
- Another exemplary dosing regimen comprises administering a dose (such as an intravenous dose) of an anti-HCV antibody as described herein to an individual receiving a liver transplant.
- the individual is administered a dose (such as an intravenous dose) of an anti-HCV antibody before receiving a liver transplant.
- the individual is administered a dose (such as an intravenous dose) of an anti-HCV antibody while receiving a liver transplant. In some embodiments, the individual is administered a dose (such as an intravenous dose) of an anti- HCV antibody after receiving a liver transplant. In some embodiments, the individual is administered a dose (such as an intravenous dose) of an anti-HCV antibody for at least one month after receiving a liver transplant.
- other dosage regimens may be useful.
- the progress of this therapy is easily monitored by conventional techniques and assays. In some embodiments, the progress of the therapy is monitored by a reduction or suppression in viral load. In some embodiments, the progress of the therapy is monitored by a reduction of ALT levels.
- an anti-HCV antibody or fragment thereof comprises a variable heavy chain region selected from the group consisting of SEQ ID NOs: 17, 18, 19, 20, 21, 22, and 23, and a variable light chain region selected from the group consisting of SEQ ID NOs:24, 25, and 26.
- the fragment of the anti-HCV antibody is selected from the group consisting of a Fab fragment, a Fab' fragment, a F(ab') 2 fragment, a scFv, a Fv, and a diabody.
- the anti-HCV antibody or fragment thereof binds to HCV.
- the anti-HCV antibody or fragment thereof is capable of binding to HCV E2 protein, soluble HCV E2 protein, or a heterodimer of HCV El protein and HCV E2 protein.
- the anti-HCV antibody or fragment thereof binds HCV E2 protein.
- the HCV E2 protein is from one or more of the HCV genotypes selected from the group consisting of genotype 1 ⁇ e.g., genotype la and genotype lb), genotype 2 ⁇ e.g., genotype 2a, genotype 2b, genotype 2c), genotype 3 ⁇ e.g., genotype 3a), genotype 4 ⁇ e.g., genotype 4a), genotype 5, and genotype 6.
- the anti-HCV antibody or fragment thereof inhibits the interaction of HCV E2 protein with CD81.
- the anti-HCV antibody or fragment thereof prevents and/or inhibits HCV entry into the cell.
- the cell is a liver cell, e.g., hepatocyte.
- an article of manufacture or kit containing anti- HCV antibodies or fragments thereof useful for the treatment, prevention and/or diagnosis of the disorders described above comprises a container and a label or package insert on or associated with the container.
- Suitable containers include, for example, bottles, vials, syringes, IV solution bags, etc.
- the containers may be formed from a variety of materials such as glass or plastic.
- the container holds a composition which is by itself or combined with another composition effective for treating, preventing and/or diagnosing the condition and may have a sterile access port (for example the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle).
- At least one active agent in the composition is an anti-HCV antibody or fragment thereof of the invention.
- the label or package insert indicates that the composition is used for treating the condition of choice.
- the article of manufacture or kit may comprise (a) a first container with a composition contained therein, wherein the composition comprises an anti-HCV antibody or fragment thereof of the invention; and (b) a second container with a composition contained therein, wherein the composition comprises a further cytotoxic or otherwise therapeutic agent.
- the article of manufacture or kit in this embodiment of the invention may further comprise a package insert indicating that the compositions can be used to treat a particular condition.
- the article of manufacture or kit may further comprise a second (or third) container comprising a pharmaceutically-acceptable buffer, such as bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's solution and dextrose solution. It may further include other materials desirable from a commercial and user standpoint, including other buffers, diluents, filters, needles, and syringes.
- BWFI bacteriostatic water for injection
- phosphate-buffered saline such as bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's solution and dextrose solution.
- BWFI bacteriostatic water for injection
- phosphate-buffered saline such as bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's solution and dextrose solution.
- BWFI bacteriostatic water for injection
- Ringer's solution such as
- kits for example, research, detection and/or diagnostic kits.
- kits typically contain the anti-HCV antibody or fragment thereof as described herein.
- the antibody is labeled or a secondary labeling reagent is included in the kit.
- the kit is labeled with instructions for performing the intended application, for example, for performing an in vivo imaging assay.
- MRCT10.1 antibody has the HVR-H1, HVR-H2, and HVR-H3 amino acid sequences found in SEQ ID NO: 13 and the HVR-L1, HVR-L2, and HVR-L3 amino acid sequences found in SEQ ID NO: 19 as disclosed in WO2009/081285, which is incorporated herein by reference in its entirety.
- GCCTCCACCAAGGGCCCATCGGTCTTCCCCCTGGCACCCTCC-3' (SEQ ID NO:40), incorporating restriction sites EcoRV and Kpnl for the light chain and BsiWl and Apal for the heavy chain.
- Heavy and light chains were ligated into a pre-digested phage display vector (phagemid) and expressed as a monovalent Fab fragment with the heavy chain fused to the Ml 3 gene-3 minor coat protein (p3).
- phagemid pre-digested phage display vector
- p3 minor coat protein
- sE2 6 6i was recombinantly expressed in SF9 insect cells, purified and biotinylated at reactive ⁇ -amino groups of lysine residues using Sulfo-NHS-SS- Biotin (Pierce) according to the manufacturer's instructions. Incubations of a phage library with the target were performed in a buffer containing 3% milk PBS/0.01 Polysorbate-20, washed with the same buffer and phage was eluted from beads using 0.1M DTT.
- Phage eluted from each round of selection were propagated for the next round by growth in XL-1 Blue cells (Agilent Technologies), Selection rounds alternated between sE2 6 6i produced from HCV genotype la or genotype lb sequences. Selected clones were ranked by competitive phage-binding as previously described in Lee et al. J Mol Biol. (2004), 340: 1073- 93.
- Anti-HCV antibody variants of humanized MRCT10.1.1 were prepared by altering specific heavy and light chain CDR residues based on affinity changes selected by phage display.
- VL-N30Y and VH-T98S were residue alterations shown to improve affinity and were used to generate V317, V361, and V362 ( Figure 2 and Table 2).
- VI and V362 a potential N-glycosylation site in heavy chain CDR-H2 was removed by mutating Asn60 to serine.
- Asn-Leu-Ser represented a motif where N-glycosylation could occur
- variants were made with SLS (e.g., VI and V362) and NPS (e.g., V6, V335, and V361) at wild type positions N60, L61, S62 according to Kabat numbering of heavy chain CDR-H2 (Table 2).
- Variants were also made by altering sites of the light chain with potential molecular instability. Specifically, an Asp-Gly motif (D27c and G27d according to Kabat numbering) with possible isomerization capability was removed by mutating CDR-L1 D27c to serine (S). Additional mutations were made to improve binding affinity to soluble E2 protein ( Figure 2, Tables 2, 3, and 4).
- HVRs Heavy chain hypervariable regions
- HVRs Light chain hypervariable regions
- a single colony was picked from growth of the transformation mixture on LB-agar plates containing 50 ⁇ g/mL carbeniciUin and was used to inoculate 1 mL of 2YT broth containing 50 ⁇ g/mL carbeniciUin.
- the culture was grown at 37 °C for 6 to 8 hours with continuous shaking, A 10 ⁇ . aliquot of 10 12 pfu/mL M13K07 helper phage (New England Biolabs) was added to the culture and incubation with shaking was continued at 37°C for 15 min before 1 mL of the culture was transferred to 50 mL of 2YT broth containing 50 ⁇ g/mL carbeniciUin and 50 ⁇ g/mL kanamycin.
- the culture was grown overnight at 37°C and the culture was subsequently centrifuged at 6,000 rpm for 10 minutes to pellet cells from the supernatant.
- the supernatant was collected and subjected to two cycles of polyethylene glycol (PEG) precipitation.
- PEG precipitation cycle consisted of resuspending the supernatant in a 1/5 volume of 20% PEG/2.5 M NaCl solution to precipitate phage particles followed by incubation for 10 minutes at room temperature, centrifugation at 13,000 rpm for 15 minutes to collect the phage pellet, re-suspension of the phage pellet in phosphate buffered saline (PBS) before centrifugation at 13,000 rpm for 15 minutes.
- PBS phosphate buffered saline
- the phage pellet was re-suspended in 1 mL PBS for preparation of ssDNA from the phage pellet using a Qiagen Ml 3 spin kit according to the manufacturer's instructions. After elution of the ssDNA from the spin column with 100 ⁇ ⁇ elution buffer, the absorbance at 260 nm was measured to determine MRCT10.1 ssDNA concentration where an A260 reading of 1 was equivalent to 33 r ⁇ g/ ⁇ L ⁇ ssDNA.
- specific oligonucleotides 330 r ⁇ g/ ⁇ L ⁇ stock concentration
- the oligonucleotides were phosphorylated by preparing a solution containing 2 ⁇ L ⁇
- oligonucleotide 2 ⁇ , ⁇ TM (0.5 M Tris-HCl pH 7.5, 0.1 M MgCl 2 ) buffer, 2 ⁇ , 10 mM rATP, 1 ⁇ , 100 mM DTT, and 12 ⁇ , H 2 0.
- a 1 ⁇ , aliquot of T4 polynucleotide kinase (New England BioLabs) was added and the solution was incubated at 37°C for 30 min.
- a 2 ⁇ L ⁇ aliquot of phosphorylated oligonucleotide was annealed with 1 ⁇ g MRCT10.1 ssDNA in a solution containing 2.5 ⁇ ⁇ ⁇ TM buffer and an amount of H 2 0 that brought the total volume to 25 ⁇ ⁇ .
- the mixture was incubated for 1 minute at 90°C then 5 minutes at 50°C before being placed on ice.
- the reaction was initiated by adding 1 ⁇ 10 mM rATP, 1 ⁇ L ⁇ 25 mM dNTP mix (equal volumes of 100 mM each dATP, dCTP, dTTP, dGTP), 1.5 ⁇ , 100 mM DTT, 0.6 ⁇ , T4 DNA ligase (New England BioLabs), 0.3 ⁇ , T7 DNA Polymerase (New England BioLabs) and the mixture was incubated at 37°C for 1.5 hours.
- a 1 ⁇ L ⁇ portion of the mutagenesis reaction was used to transform XL- 1 Blue (Stratagene) using the manufacturer's protocol and the transformants were plated on LB agar plates containing 50 ⁇ g/mL carbeniciUin.
- dsDNA was prepared using a Qiagen mini-prep spin kit (Qiagen) and the correct sequence clones were identified using dideoxynucleotide sequencing.
- variable domains of the generated variants were sub-cloned into pRK5 vectors for transient mammalian cell expression.
- Variable regions were amplified by PCR using oligonucleotides: forward primer 5'- CTCGGTTCTATCG ATTG A ATTCC ACC ATGGG- 3 ' (SEQ ID NO:59) and reverse primer 5 ' -GCC AAGGGACCACGGTC ACCGTCTCCTC AG-3 ' (SEQ ID NO:60) for heavy chain and forward primer 5'- GGAGTACATTCAGATATCGTGTTGACGC-3 ' (SEQ ID NO:61) and reverse primer 5'- CTGGAGATCAAACGGACCGTGGCTGCAC -3' (SEQ ID NO:62) for light chain to incorporate restriction sites for directional cloning.
- VL Light chain variable domains
- VH Heavy chain variable domains
- Digested variable region fragments were ligated into pre-digested pRK5 vectors encoding human kappa I antibody for the light chain, or human IgGl antibody for the heavy chain, and the ligation product was transformed into Escherichia coli strain XL 1 -blue (Strategene, La Jolla, CA, USA). Single colonies were selected for plasmid purification. Correctly subcloned plasmids were determined by DNA sequencing.
- transfection mixture A 1 mL aliquot of transfection mixture was added to each T150 flask and the cells were incubated. After incubation, conditioned media was collected 4 to 7 days post-transfection. PMSF and bovine lung aprotinin were added to final concentrations of 1 mM and 1.2 ⁇ g/mL, respectively. The media was filtered using a low protein binding polystyrene bottle with a 0.22 ⁇ cellulose acetate filter to remove detached cells. IgG was purified on a 0.5 to 1 mL of rProtein A agarose column.
- the generated anti-HCV antibodies were compared to wild- type MRCT10.1 for affinity.
- surface plasmon resonance (SPR) measurements on a Biacore A 100 instrument (GE Healthcare Biosciences AB) was used to measure kinetics of antibody variants binding to soluble E2 extracellular domain.
- SPR surface plasmon resonance
- IgG variants were captured in each automated cycle with a contact time of 3 minutes at a flow rate of ⁇ /min.
- Soluble E2 antigen was injected for 2 minutes at a flow rate of 20 ⁇ 1/ ⁇ in a concentration series ranging from 200nM to 0.78nM in 2-fold increments.
- Four flow cells were utilized simultaneously to capture antibody on spots 1 and 5, with spots 2 and 4 reserved as internal references containing only the capturing IgG.
- V362 3.3 + 0.5 0.9 + 0.1 2.7 + 0.5 3.6 + 0.4 0.9 + 0.1 2.6 + 0.5
- HCVpp HCV pseudoparticles
- HEK 293T cells were co-transfected with the Delta 8.9 plasmid containing gag-pol, the CMV-Luc-DsRed lentiviral transfer construct, and a plasmid expressing HCV E1E2 sequences from genotype 2a (J6CF) using Lipofectamine 2000, according to the manufacturer's instructions. Forty-eight hours following transfection, the medium containing HCVpp was collected, clarified, filtered through 0.45- ⁇ pore-sized membranes and used for infection of Huh-7.5 cells. HCVpp stocks were normalized for luciferase activity before infecting cells. Neutralization assays using HCVpp viruses were performed in 96-well plates (Costar).
- Values represent average EC 90 values + SEM and are representative of at least two
- Anti-HCV antibodies were also assayed for neutralization of infectious cell culture HCV (HCVcc) of wild- type genotype 2a Jcl in addition to E2 mutants of genotype 2a Jcl that arise when treated with other anti-HCV antibodies that target the same epitope of the E2 protein.
- HCVcc neutralization assay Jcl HCVcc virus stocks were generated by transfecting Huh7.5 cells with in vitro transcribed full-length HCV RNA as previously described in Zhong et al., PNAS, (2005), 102:9294-9, which is incorporated herein by reference in its entirety.
- genotype 2a Jcl genome were chemically synthesized using sequences genotype 2a JFH-1 (GenBank accession number AB047639) and genotype 2a J6CF (GenBank accession number AF177036) based on the chimeric strategy as described in Pietschmann et al., PNAS, (2006), 103:7408-7413 and cloned into pUC19 plasmid (pUC Jcl). Plasmids were linearized with Xbal and in vitro transcribed RNA was generated using Megascript kit (Ambion) according to the manufacturer's instructions.
- HCV RNA was transfected into Huh-7.5 cells by electroporation and supernatants from Jcl RNA-transfected cells were harvested starting from 3 days post transfection, titered and pooled to generate an HCVcc stock.
- HCVcc neutralization assays were performed as described for the HCVpp neutralization assay above.
- Binding of V362 to either E2 41 4 J peptide or full-length E1E2 expressed in transfected cells was assayed. Binding of Fab fragments to chemically synthesized peptides (non-glycosylated) was measured by Surface plasmon resonance (SPR) or Bio-Layer
- V362 showed measurable binding affinity to the E2 412-423 peptide epitope with a Ku of 30 nM. Ratios of binding alanine mutant versus wild-type versions of the peptide were measured to further characterize binding of V362 to the E2 epitope ( Figure 3 A). V362 did not bind the W420A mutant peptide but did bind wild-type and N417A mutant peptides ( Figure 3A). Mutants L413A and G418A affected V362 binding by ⁇ 8-fold and ⁇ 2- fold, respectively, and binding of V362 to N415A only decreased by 2.8-fold ( Figure 3A).
- Example 4 Combination of anti-HCV antibodies with HCV antivirals enhanced suppression of infection and spread of resistant HCV.
- the N417S resistant mutant genotype 2a HCVcc rapidly emerges in vitro in the presence of previously described AP33 antibody.
- the ability of anti-HCV antibodies when used in combination with an HCV direct acting antiviral (DAA) to enhance antiviral efficacy and suppress the emergence of resistant HCVcc was assessed.
- DAA direct acting antiviral
- V362 antibody could suppress the emergence of resistance to an approved HCV NS3 protease inhibitor (Telaprevir)
- DMSO-differentiated Huh-7.5 cells were infected with Jcl HCVcc alone or in the presence of Telaprevir, V362 antibody or a combination of both Telaprevir and V362 antibody ( Figure 4).
- Telaprevir or a combination of 2 ⁇ Telaprevir and 10 ⁇ g/ml V362 antibody.
- DMSO-differentiated Huh-7.5 cells were infected with Jcl HCVcc alone or in the presence of Telaprevir and IFN-a or a combination of Telaprevir, IFN-a and V335 antibody (Figure 5A).
- V335 antibody was more effective at suppressing viral infection and spread in vitro as compared to MRCT10.1 antibody ( Figure 5B, inverted filled diamonds).
Abstract
This invention relates to compositions comprising anti-HCV antibodies and methods of inhibiting HCV infection and treating HCV-associated disease with anti-HCV antibodies, as well as articles of manufacture comprising anti-HCV antibodies.
Description
ANTI-HCV ANTIBODIES AND METHODS OF USING THEREOF
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of U.S. Provisional Patent Application Serial No. 61/755,932, filed January 23, 2013, and U.S. Provisional Patent Application Serial No.
61/770,272, filed February 27, 2013, each of which is incorporated herein by reference in its entirety.
SUBMISSION OF SEQUENCE LISTING ON ASCII TEXT FILE
[0002] The content of the following submission on ASCII text file is incorporated herein by reference in its entirety: a computer readable form (CRF) of the Sequence Listing (file name: 146392016540SEQLIST.txt, date recorded: January 21, 2014, size: 33,880 bytes).
FIELD OF THE INVENTION
[0003] The present invention relates to anti-HCV antibodies capable of inhibiting HCV infection and compositions comprising these antibodies, as well as methods for producing and using the same.
BACKGROUND
[0004] Hepatitis C virus (HCV), a member of the Flaviviridae family of viruses, is a major cause of liver disease such as chronic hepatitis and hepatocellular carcinoma (HCC). HCV is most commonly transmitted by direct blood contact, for instance by needle stick injuries in healthcare settings, injection drug use, receipt of contaminated blood transfusion and from mother to infant transmission. About 75- 85% of infected people develop chronic HCV infection, with 60-70% of chronically infected people developing chronic liver disease which can progress to cirrhosis and HCC. It is estimated that about 170 million people worldwide have been infected with HCV and 350,000 infected people die annually from HCV- associated liver disease.
[0005] Currently, there is no approved vaccine against HCV. The recommended treatment for chronic infection involves a 48-week course of peginterferon-a-2b (PegIFN-a-2b) or -a- 2a (PegIFN-a-2a) combined with ribarvirin (RBV). This standard of care (SoC) of PEG-
IFNa plus RBV is of limited success, offering a 35-40% cure rate depending on the virus genotype, and is associated with side effects such as flu-like symptoms, fatigue, anemia and severe depression. As a result, a large number of patients forgo treatment. New Direct Acting Antiviral (DAA) therapies, such as viral protease and viral polymerase inhibitors, are currently under development for use as monotherapy or combination therapy and have the potential to replace the current SoC. However, due to the high mutational rate and therefore high genetic variability in HCV, effectiveness of such DAA therapies can be genotype specific and as a result such treatments are limited to subpopulations of HCV infected individuals. Six major HCV genotypes have been identified that are further divided into at least 70 different subtypes. Currently, HCV genotype 1 represents greater than 70% of infections in the US, Europe and Australia, and is the most difficult to cure. A significant challenge for the development of vaccines is the identification of protective epitopes that are conserved in the majority of viral genotypes and subtypes. This problem is compounded by the fact that the envelope proteins, the natural target for the neutralizing response, are two of the most variable proteins.
[0006] HCV contains a positive-strand RNA genome which encodes a single polyprotein of approximately 3000 amino acids in length that is post-translationally processed to produce at least ten different proteins: core, envelope proteins El and E2, p7, and non- structural proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B. HCV entry into hepatocytes occurs through the coordinated interactions between the HCV E1-E2 heterodimer and at least four essential host cellular factors; CD81, scavenger receptor B type I (SR-BI), occludin (OCLN) and claudin 1 (CLDNl). In particular, the HCV E2 glycoprotein binds CD81 and SR-BI. In HCV genotype la strain H77c, the E2 glycoprotein extends from amino acids 384 to 746 of the polyprotein and has regions of extreme variability. An immunodominant portion of E2 protein, located within the N-terminal 27 residues (amino acids 384 to 411) involved in virus binding and entry, also contains the most variable region (also known as the first hypervariable region) and therefore is challenging to target with antibodies that recognize conserved
conformational epitopes. Antibodies specific for epitopes within the first hypervariable region have been reported to inhibit the binding of E2 glycoprotein to cells and to block HCV infectivity in vitro and in vivo. A region immediately downstream of the first hypervariable region contains an epitope encompassing residues 412-423 of E2 protein. Antibodies targeting this epitope inhibit the interaction between CD81 and a range of presentations of E2 protein, including soluble E2, E1E2 and virus-like particles. See Owsianka et al., J. Gen. Virol., (2001), 82(Pt 8): 1877-83; WO2006/ 100449; and WO2009/081285. However,
antibodies with improved characteristics that make them amenable for clinical use in monotherapy and combination therapy are still needed.
[0007] All references cited herein, including patent applications and publications, are hereby incorporated by reference in their entirety.
BRIEF SUMMARY OF THE INVENTION
[0008] The invention provided herein discloses, inter alia, anti-HCV antibodies capable of inhibiting HCV infection and compositions comprising these antibodies, as well as methods for producing and using these antibodies for the treatment or prevention of HCV-associated disorders or disease. In some embodiments, an HCV-associated disorder or disease is HCV infection. In some embodiments, an HCV-associated disorder or disease is liver disease.
[0009] Accordingly, in one aspect, the invention provides an isolated antibody that binds hepatitis C virus E2 protein comprising a heavy chain variable region and a light chain variable region, (a) wherein the heavy chain variable region comprises three hypervariable region (HVR) sequences, wherein (i) HVR-Hl comprises GXiSX2TSGYWN (SEQ ID NO: l), wherein is D or E, and X2 is I or L; (ii) HVR-H2 comprises
YISYSGSTYYXiX2SLRS (SEQ ID NO:2), wherein Χ is N or S, and X2 is P or L; and (iii) HVR-H3 comprises ALITTX \ T Y AMD Y (SEQ ID NO:3), wherein Xi is S or T; and (b) wherein the light chain variable region comprises three HVR sequences, wherein (iv) HVR- Ll comprises RASES VXiGYGX2SFLH (SEQ ID NO:4), wherein Xiis D or S, and X2= N or Y; (v) HVR-L2 comprises LASNLNS (SEQ ID NO:5); and (vi) HVR-L3 comprises
QQNNVDPWT (SEQ ID NO:6); wherein the antibody is not an antibody comprising (a) HVR-Hl comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 11 ; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 15; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising an amino acid sequence selected from SEQ ID NO:6. In some embodiments, the antibody comprises HVR-Hl comprising the amino acid sequence of SEQ ID NO:7 or 8, HVR-H2 comprising the amino acid sequence of SEQ ID NO:9, 10 or 11, and HVR-H3 comprising the amino acid sequence of SEQ ID NO: 12 or 13. In some embodiments herein, the antibody comprises HVR-L1 comprising the amino acid sequence of SEQ ID NO: 14, 15 or 16, HVR- L2 comprising the amino acid sequence of SEQ ID NO:5, and HVR-L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the antibody comprises (a)
HVR-H1 comprising the amino acid sequence of SEQ ID NO:7, HVR-H2 comprising the amino acid sequence of SEQ ID NO:9 or 10, and HVR-H3 comprising the amino acid sequence of SEQ ID NO: 12; and/or (b) HVR-Ll comprising the amino acid sequence of SEQ ID NO: 14, HVR-L2 comprising the amino acid sequence of SEQ ID NO:5, and HVR-L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the antibody comprises the heavy chain variable region sequence of SEQ ID NO: 17, 18, or 22, and/or the light chain variable region sequence of SEQ ID NO:24. In some embodiments, the antibody comprises the heavy chain variable region sequence of SEQ ID NO: 19, 20, 22, or 23, and/or the light chain variable region sequence of SEQ ID NO:24 or 25. In some embodiments, the antibody comprises the heavy chain variable region sequence of SEQ ID NO:21, and/or the light chain variable region sequence of SEQ ID NO:24. In some embodiments, the antibody comprises the heavy chain sequence of SEQ ID NO:27 or 28, and the light chain sequence of SEQ ID NO:30. In some embodiments herein, the antibody is a humanized antibody. In some embodiments herein, the antibody is an antigen-binding fragment. In some
embodiments herein, the antibody is an antibody fragment selected from the group consisting of a Fab, Fab'-SH, Fv, scFv, and (Fab')2 fragment. In some embodiments herein, the antibody inhibits infection of one or more HCV genotypes (such as genotype la, genotype lb, genotype 2a, and genotype 4a) to liver cells.
[0010] In another aspect, the invention provides an isolated antibody that binds hepatitis C virus E2 protein comprising a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising amino acid sequence SGYWN (SEQ ID NO:33), CDR H2 comprising the amino acid sequence of SEQ ID NO:9, 10, or 11, CDR H3 comprising amino acid sequence ITTSTYAMDY (SEQ ID NO:34) or ITTTTYAMDY (SEQ ID NO:35); and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, 15, or 16, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the anti-HCV antibody is not an antibody comprising a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the antibody comprises a heavy
chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO:9 or 10, CDR H3 comprising the amino acid sequence of SEQ ID NO:34; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments herein, the antibody is a humanized antibody. In some embodiments herein, the antibody is an antigen-binding fragment. In some embodiments herein, the antibody is an antibody fragment selected from the group consisting of a Fab, Fab'-SH, Fv, scFv, and (Fab')2 fragment. In some embodiments herein, the antibody inhibits infection of one or more HCV genotypes (such as genotype la, genotype lb, genotype 2a, and genotype 4a) to liver cells.
[0011] In another aspect, the invention provides an antibody that binds hepatitis C virus E2 protein produced by a method comprising culturing a host cell comprising a nucleic acid encoding any of the antibodies disclosed herein under a condition suitable for expression of the nucleic acid; and recovering the antibody produced by the cell. In some embodiments, the host cell comprises a nucleic acid encoding a VL amino acid sequence, and/or a nucleic acid encoding a VH amino acid sequence of any of the anti-HCV antibodies described above and herein.
[0012] In yet another aspect, the invention provides a pharmaceutical composition comprising any of the antibodies disclosed herein, and a pharmaceutically acceptable carrier. In yet another aspect, the invention provides a kit or an article of manufacture comprising one or more of the anti-HCV antibodies described above and herein. In some embodiments, the kit or article of manufacture may further comprise a label or package insert indicating that the antibody is used for the treatment, prevention and/or diagnosis of the disorders described herein (e.g., hepatitis C virus infection). In some embodiments, the kit or article of manufacture may further comprise a second therapeutic agent.
[0013] In another aspect, the invention also provides an isolated nucleic acid encoding any of the antibodies disclosed herein. In still another aspect, the invention provides a vector comprising a nucleic acid encoding any of the antibodies disclosed herein. In a further embodiment, the vector is an expression vector. In some embodiments, the vector comprises a nucleic acid encoding a VL amino acid sequence of any of the anti-HCV antibodies described above and herein. In some embodiments, the vector comprises a nucleic acid
encoding a VH amino acid sequence of any of the anti-HCV antibodies described above and herein.
[0014] In one aspect, the invention also provides a host cell comprising a nucleic acid encoding any of the antibodies disclosed herein. In a further embodiment, the host cell is prokaryotic or eukaryotic. In some embodiments, the host cell comprises a nucleic acid encoding a VL amino acid sequence, and/or a nucleic acid encoding a VH amino acid sequence of any of the anti-HCV antibodies described above and herein.
[0015] Accordingly, in another aspect, the invention provides a method of producing an antibody comprising culturing a host cell comprising a nucleic acid encoding any of the antibodies disclosed herein under a condition suitable for expression of the nucleic acid; and recovering the antibody produced by the cell. In some embodiments, the method further comprises purifying the antibody. In some embodiments, the host cell comprises a nucleic acid encoding a VL amino acid sequence, and/or a nucleic acid encoding a VH amino acid sequence of any of the anti-HCV antibodies described above and herein.
[0016] In another aspect, the invention provides a method for treating or preventing a hepatitis C virus infection in a subject, comprising administering to the subject an effective amount of any of the antibodies disclosed herein. In some embodiments, the subject is a human. In some embodiments, the subject has been diagnosed with the hepatitis C virus infection. In some embodiments, the hepatitis C virus infection is an acute hepatitis C virus infection. In some embodiments, the hepatitis C virus infection is a chronic hepatitis C virus infection. In some embodiments, treating the hepatitis C virus infection comprises reducing viral load. In another embodiment, the method further comprises administering a second therapeutic agent. In a further embodiment, the second therapeutic agent is a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a cyclophilin inhibitor, an antibody that targets a non-E2 HCV protein, or a combination thereof. In another further embodiment, the second therapeutic agent is a HCV protease inhibitor selected from the group consisting of Teleprevir, Boceprevir, Semeprevir (TMC435), BI201335, Danoprevir/r (RG7227), Vaniprevir (MK-7009), Narlaprevir (SCH 900518), Asunaprevir, GS-9256, GS-9451, ABT-450/r, ACH-1625, ACH-2684, and MK-5172. In yet another further embodiment, the second therapeutic agent is a polymerase inhibitor selected from the group consisting of PSI-7977, Mercitabine, IDX184, PSI-938, INX-189, Tegobuvir, Filibuvir, Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055. In some
embodiments herein, the treatment is an interferon-free treatment. In some embodiments herein, the subject has sustained virologic response for at least 12 weeks after stopping the
treatment. In some embodiments herein, the viral load in the subject has been reduced to an undetectable level after the treatment. In some embodiments herein, the viral resistance in the subject is undetectable or low. In some embodiments herein, the subject is not responsive to an interferon treatment.
[0017] In another aspect, the invention provides a method for treating or preventing a hepatitis C virus infection in a subject, comprising administering to the subject an effective amount of an antibody that specifically binds hepatitis C virus E2 protein and a second therapeutic agent. In another aspect, the invention provides a method of preventing developing resistance to treatment, comprising administering an effective amount of an antibody that specifically binds hepatitis C virus E2 protein and a second therapeutic agent. In some embodiments, the second therapeutic agent is a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a cyclophilin inhibitor, an antibody that targets a non-E2 HCV protein, and a combination thereof. In some embodiments, the antibody comprises a heavy chain variable region and a light chain variable region, (a) wherein the heavy chain variable region comprises three hypervariable region (HVR) sequences, wherein (i) HVR-H1 comprises GXiSX2TSGYWN (SEQ ID NO: l), wherein Χ is D or E, and X2 is I or L; (ii) HVR-H2 comprises YISYSGSTYYXiX2SLRS (SEQ ID NO:2), wherein X1 is N or S, and X2 is P or L; and (iii) HVR-H3 comprises
ALITTX i TY AMD Y (SEQ ID NO:3), wherein Χ is S or T; and (b) wherein the light chain variable region comprises three HVR sequences, wherein (iv) HVR-L1 comprises
RASES VX1GYGX2SFLH (SEQ ID NO:4), wherein Xjis D or S, and X2= N or Y; (v) HVR- L2 comprises LASNLNS (SEQ ID NO:5); and (vi) HVR-L3 comprises QQNNVDPWT (SEQ ID NO:6). Any of the anti-HCV antibodies described above and herein may be administered to the subject for the treatment or prevention.
[0018] In yet another aspect, the invention provides a method of preventing of HCV infection of a transplanted liver, comprising administering to the subject an effective amount of any of the antibodies disclosed herein before, during or after the subject receives the liver transplant.
[0019] The invention also provides use of an anti-HCV antibody described above and herein in the manufacture of a medicament for use in any of the methods described above and herein (e.g., for treating or preventing a hepatitis C virus infection in a subject, for preventing developing resistance to treatment, or preventing HCV infection of a transplanted liver). The invention also provides an anti-HCV antibody described above and herein for use in any of the methods described above and herein (e.g., for treating or preventing a hepatitis C virus
infection in a subject, for preventing developing resistance to treatment, or preventing HCV infection of a transplanted liver).
[0020] It is to be understood that one, some, or all of the properties of the various embodiments described herein may be combined to form other embodiments of the present invention. These and other aspects of the invention will become apparent to one of skill in the art.
BRIEF DESCRIPTION OF THE DRAWINGS
[0021] Figure 1 is a diagram of the HCV E2 protein epitope binding site.
[0022] Figure 2 shows an amino acid sequence alignment of the A) light chain variable region and B) heavy chain variable region of MRCT10.1, V361, and V362 numbered according to Kabat. Boxed amino acid sequences indicate hypervariable regions (HVRs). Bars over the sequence indicate the complementarity determining regions (CDRs). From the N-terminus to C-terminus (left to right), amino acid sequences outside of boxed HVRs indicate FR1, FR2, FR3, and FR4. "MRCT10" in the figure refers to the MRCT10.1 amino acid sequence.
[0023] Figure 3 is a series of graphs showing binding of V362 to wild-type (WT) and
412-423
mutant soluble E2 proteins (sE266i) and E2 " peptides. A) Ratio of dissociation constants (KD) for V362 binding to alanine mutant peptides relative to WT peptide
4i2QLINTNGSWHIN423GSGK-biotin (E2412-423-biotin) (SEQ ID NO:36). Asterisk denotes no detectable binding between W420A mutant peptide and V362. B) Binding of AP33 and V362 neutralizing antibodies to lysates of untransfected 293 cells (Cells) or 293 cells transfected with genotype 2a (J6CF) HCV ElE2-expressing plasmids containing various mutations at N415 or N417. Representative data from three independent experiments. (C and D) Biacore sensorgrams demonstrating binding of MRCT10.1 Fab to WT C) or N417S
412 423
D) E2"1 ^J peptide. The Fab series was diluted two-fold starting from 1000 nM down to 15.6 nM. MRCT10.1 Fab bound to N417S E2412"423 peptide with faster off-rate compared to WT.
[0024] Figure 4 is a graph depicting enhancement of the antiviral effect of an NS3 protease inhibitor, Telaprevir, with an anti-HCV antibody. DMSO-differentiated Huh-7.5 cells were infected with Jcl HCVcc (MOI=0.05) alone (filled squares) or in the presence V362 (10 μg/mL, open circles), Telaprevir (2 μΜ, open triangles) or a combination of 10 μg/mL V362 and 2 μΜ Telaprevir (open diamonds). HCV RNA copies were measured at the indicated times post infection. HCV cDNA derived from the day 22 post-infection cultures was
sequenced. Dotted line represents the limit of qPCR linear range. Representative data from two independent experiments.
[0025] Figure 5 is a series of graphs depicting enhancement of the antiviral effect of Telaprevir and interferon combination treatment with anti-HCV antibodies. A) DMSO- differentiated Huh-7.5 cells were infected with Jcl HCVcc (MOI=0.05) alone (filled squares) or in the presence 0.3 μΜ Telaprevir plus 5 IU/ml Interferon alpha (IFN-a) (open circles) or a combination of 0.3 μΜ Telaprevir and 5 IU/ml IFN-a plus V335 (10 μg/mL, filled inverted diamonds). Asterisks denote undetectable HCV cDNA in cultures treated with a combination of V335, Telaprevir, and IFN-a at 10, 14, 22, and 30 days post infection. Representative data from two independent experiments. Dotted line represents the limit of qPCR linear range. B) DMSO-differentiated Huh-7.5 cells were infected with Jcl HCVcc (MOI=0.05) alone (filled squares) or in the presence 0.3 μΜ Telaprevir plus 5 IU/ml Interferon alpha (IFN-a) (open circles) or a combination of 0.3 μΜ Telaprevir and 5 IU/ml IFN-a plus MRCT10.1 (10 μg/mL, filled inverted diamonds). HCV RNA copies were measured at various times post infection by RT-qPCR. Representative data from two independent experiments. Dotted line represents the limit of qPCR linear range.
DETAILED DESCRIPTION OF THE INVENTION
[0026] Provided herein are antibodies that bind HCV E2 protein (hereinafter referred to as "anti-HCV antibodies") that inhibit HCV cellular entry and HCV infection. Anti-HCV antibodies of the present invention can be used as a therapeutic agent for use in the treatment of HCV-associated diseases.
I. General techniques
[0027] The techniques and procedures described or referenced herein are generally well understood and commonly employed using conventional methodology by those skilled in the art, such as, for example, the widely utilized methodologies described in Sambrook et al., Molecular Cloning: A Laboratory Manual 3d edition (2001) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.; Current Protocols in Molecular Biology (F.M. Ausubel, et al. eds., (2003)); the series Methods in Enzymology (Academic Press, Inc.): PCR 2: A
Practical Approach (M.J. MacPherson, B.D. Hames and G.R. Taylor eds. (1995)), Harlow and Lane, eds. (1988) Antibodies, A Laboratory Manual, and Animal Cell Culture (R.I.
Freshney, ed. (1987)); Oligonucleotide Synthesis (M.J. Gait, ed., 1984); Methods in
Molecular Biology, Humana Press; Cell Biology: A Laboratory Notebook (J.E. Cellis, ed.,
1998) Academic Press; Animal Cell Culture (R.I. Freshney), ed., 1987); Introduction to Cell and Tissue Culture (J. P. Mather and P.E. Roberts, 1998) Plenum Press; Cell and Tissue Culture: Laboratory Procedures (A. Doyle, J.B. Griffiths, and D.G. Newell, eds., 1993-8) J. Wiley and Sons; Handbook of Experimental Immunology (D.M. Weir and C.C. Blackwell, eds.); Gene Transfer Vectors for Mammalian Cells (J.M. Miller and M.P. Calos, eds., 1987); PCR: The Polymerase Chain Reaction, (Mullis et al., eds., 1994); Current Protocols in Immunology (J.E. Coligan et al., eds., 1991); Short Protocols in Molecular Biology (Wiley and Sons, 1999); Immunobiology (C.A. Janeway and P. Travers, 1997); Antibodies (P. Finch, 1997); Antibodies: A Practical Approach (D. Catty., ed., IRL Press, 1988-1989); Monoclonal Antibodies: A Practical Approach (P. Shepherd and C. Dean, eds., Oxford University Press, 2000); Using Antibodies: A Laboratory Manual (E. Harlow and D. Lane (Cold Spring Harbor Laboratory Press, 1999); The Antibodies (M. Zanetti and J. D. Capra, eds., Harwood
Academic Publishers, 1995); and Cancer: Principles and Practice of Oncology (V.T. DeVita et al., eds., J.B. Lippincott Company, 1993).
II. Definitions
[0028] An "acceptor human framework" for the purposes herein is a framework comprising the amino acid sequence of a light chain variable domain (VL) framework or a heavy chain variable domain (VH) framework derived from a human immunoglobulin framework or a human consensus framework, as defined below. An acceptor human framework "derived from" a human immunoglobulin framework or a human consensus framework may comprise the same amino acid sequence thereof, or it may contain amino acid sequence changes. In some embodiments, the number of amino acid changes are 10 or less, 9 or less, 8 or less, 7 or less, 6 or less, 5 or less, 4 or less, 3 or less, or 2 or less. In some embodiments, the VL acceptor human framework is identical in sequence to the VL human immunoglobulin framework sequence or human consensus framework sequence.
[0029] "Affinity" refers to the strength of the sum total of noncovalent interactions between a single binding site of a molecule (e.g., an antibody) and its binding partner (e.g., an antigen). Unless indicated otherwise, as used herein, "binding affinity" refers to intrinsic binding affinity which reflects a 1: 1 interaction between members of a binding pair (e.g., antibody and antigen). The affinity of a molecule X for its partner Y can generally be represented by the dissociation constant (Kd). Affinity can be measured by common methods known in the art, including those described herein. Specific illustrative and exemplary embodiments for measuring binding affinity are described in the following.
[0030] An "affinity matured" antibody refers to an antibody with one or more alterations in one or more hypervariable regions (HVRs), compared to a parent antibody which does not possess such alterations, such alterations resulting in an improvement in the affinity of the antibody for antigen.
[0031] The terms "anti-HCV antibody", "an antibody that binds to HCV", and "an antibody that binds to HCV E2 protein" as used herein refer to an antibody that is capable of binding to HCV E2 protein with sufficient affinity such that the antibody is useful as a diagnostic and/or therapeutic agent in targeting HCV E2 protein and/or HCV. In one embodiment, the extent of binding of an anti-HCV antibody to an unrelated, non-HCV E2 protein is less than about 10% of the binding of the antibody to HCV E2 protein and/or HCV as measured, e.g., by a radioimmunoassay (RIA). In certain embodiments, an antibody that binds to HCV E2 protein and/or HCV has a dissociation constant (Kd) of < ΙμΜ, < 100 nM, < 10 nM, < 1 nM, < 0.1 nM, < 0.01 nM, or < 0.001 nM (e.g. 10"8 M or less, e.g. from 10"8 M to 10"13 M, e.g., from 10"
9 M to 10 -"13 M). In certain embodiments, an anti-HCV antibody binds to an epitope of HCV E2 protein that is conserved among HCV from different genotypes.
[0032] The term "antibody" herein is used in the broadest sense and encompasses various antibody structures, including but not limited to monoclonal antibodies, polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired antigen-binding activity.
[0033] An "antibody fragment" refers to a molecule other than an intact antibody that comprises a portion of an intact antibody that binds the antigen to which the intact antibody binds. Examples of antibody fragments include but are not limited to Fv, Fab, Fab', Fab'-SH, F(ab')2; diabodies; linear antibodies; single-chain antibody molecules (e.g. scFv); and multispecific antibodies formed from antibody fragments.
[0034] An "antibody that binds to the same epitope" as a reference antibody refers to an antibody that blocks binding of the reference antibody to its antigen in a competition assay by 50% or more, and conversely, the reference antibody blocks binding of the antibody to its antigen in a competition assay by 50% or more.
[0035] The term "chimeric" antibody refers to an antibody in which a portion of the heavy and/or light chain is derived from a particular source or species, while the remainder of the heavy and/or light chain is derived from a different source or species.
[0036] The "class" of an antibody refers to the type of constant domain or constant region possessed by its heavy chain. There are five major classes of antibodies: IgA, IgD, IgE, IgG, and IgM, and several of these may be further divided into subclasses (isotypes), e.g., IgG1;
IgG2, IgG3, IgG4, IgA1 ; and IgA2. The heavy chain constant domains that correspond to the different classes of immunoglobulins are called α, δ, ε, γ, and μ, respectively.
[0037] The term "cytotoxic agent" as used herein refers to a substance that inhibits or prevents a cellular function and/or causes cell death or destruction. Cytotoxic agents include, but are not limited to, radioactive isotopes (e.g., At211, 1131, 1125, Y90, Re186, Re188, Sm153,
212 32 212
Bi , P , Pb and radioactive isotopes of Lu); chemotherapeutic agents or drugs (e.g., methotrexate, adriamicin, vinca alkaloids (vincristine, vinblastine, etoposide), doxorubicin, melphalan, mitomycin C, chlorambucil, daunorubicin or other intercalating agents); growth inhibitory agents; enzymes and fragments thereof such as nucleolytic enzymes; antibiotics; toxins such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, including fragments and/or variants thereof; and the various antitumor or anticancer agents disclosed below.
[0038] "Effector functions" refer to those biological activities attributable to the Fc region of an antibody, which vary with the antibody isotype. Examples of antibody effector functions include: Clq binding and complement dependent cytotoxicity (CDC); Fc receptor binding; antibody-dependent cell-mediated cytotoxicity (ADCC); phagocytosis; down regulation of cell surface receptors (e.g. B cell receptor); and B cell activation.
[0039] An "effective amount" of an agent, e.g., a pharmaceutical formulation, refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired therapeutic or prophylactic result. An effective amount can be provided in one or more administrations.
[0040] A "therapeutically effective amount" is at least the minimum concentration required to effect a measurable improvement of a particular disorder (e.g., HCV infection). A therapeutically effective amount herein may vary according to factors such as the disease state, age, sex, and weight of the patient, and the ability of the anti-HCV antibody to elicit a desired response in the individual. A therapeutically effective amount is also one in which any toxic or detrimental effects of the anti-HCV antibody are outweighed by the
therapeutically beneficial effects. A "prophylactically effective amount" refers to an amount effective, at the dosages and for periods of time necessary, to achieve the desired prophylactic result. Typically but not necessarily, since a prophylactic dose is used in subjects prior to or at an earlier stage of disease, a prophylactically effective amount may be less than a therapeutically effective amount.
[0041] The term "Fc region" herein is used to define a C-terminal region of an immunoglobulin heavy chain, including native- sequence Fc regions and variant Fc regions. Although the boundaries of the Fc region of an immunoglobulin heavy chain might vary, the human IgG heavy-chain Fc region is usually defined to stretch from an amino acid residue at position Cys226, or from Pro230, to the carboxyl-terminus thereof. The C-terminal lysine (residue 447 according to the EU numbering system) of the Fc region may be removed, for example, during production or purification of the antibody, or by recombinantly engineering the nucleic acid encoding a heavy chain of the antibody. Accordingly, a composition of intact antibodies may comprise antibody populations with all K447 residues removed, antibody populations with no K447 residues removed, and antibody populations having a mixture of antibodies with and without the K447 residue. Suitable native- sequence Fc regions for use in the antibodies of the invention include human IgGl, IgG2, IgG3 and IgG4. Unless otherwise specified herein, numbering of amino acid residues in the Fc region or constant region is according to the EU numbering system, also called the EU index, as described in Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD, 1991.
[0042] "Framework" or "FR" refers to variable domain residues other than hypervariable region (HVR) residues. The FR of a variable domain generally consists of four FR domains: FR1, FR2, FR3, and FR4. Accordingly, the HVR and FR sequences generally appear in the following sequence in VH (or VL): FR1-H1(L1)-FR2-H2(L2)-FR3-H3(L3)-FR4.
[0043] The terms "full length antibody," "intact antibody," and "whole antibody" are used herein interchangeably to refer to an antibody having a structure substantially similar to a native antibody structure or having heavy chains that contain an Fc region as defined herein.
[0044] The terms "host cell," "host cell line," and "host cell culture" are used
interchangeably and refer to cells into which exogenous nucleic acid has been introduced, including the progeny of such cells. Host cells include "transformants" and "transformed cells," which include the primary transformed cell and progeny derived therefrom without regard to the number of passages. Progeny may not be completely identical in nucleic acid content to a parent cell, but may contain mutations. Mutant progeny that have the same function or biological activity as screened or selected for in the originally transformed cell are included herein.
[0045] A "human antibody" is one which possesses an amino acid sequence which corresponds to that of an antibody produced by a human or a human cell or derived from a non-human source that utilizes human antibody repertoires or other human antibody-
encoding sequences. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen-binding residues.
[0046] A "human consensus framework" is a framework which represents the most commonly occurring amino acid residues in a selection of human immunoglobulin VL or VH framework sequences. Generally, the selection of human immunoglobulin VL or VH sequences is from a subgroup of variable domain sequences. Generally, the subgroup of sequences is a subgroup as in Kabat et al., Sequences of Proteins of Immunological Interest, Fifth Edition, NIH Publication 91-3242, Bethesda MD (1991), vols. 1-3. In one embodiment, for the VL, the subgroup is subgroup kappa I as in Kabat et al., supra. In one embodiment, for the VH, the subgroup is subgroup III as in Kabat et al., supra.
[0047] A "humanized" antibody refers to a chimeric antibody comprising amino acid residues from non-human HVRs and amino acid residues from human FRs. In certain embodiments, a humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the HVRs (e.g., CDRs) correspond to those of a non-human antibody, and all or substantially all of the FRs correspond to those of a human antibody. A humanized antibody optionally may comprise at least a portion of an antibody constant region derived from a human antibody. A "humanized form" of an antibody, e.g., a non-human antibody, refers to an antibody that has undergone humanization.
[0048] The term "hypervariable region" or "HVR," as used herein, refers to each of the regions of an antibody variable domain which are hypervariable in sequence and/or form structurally defined loops ("hypervariable loops"). Generally, native four-chain antibodies comprise six HVRs; three in the VH (HI, H2, H3), and three in the VL (LI, L2, L3). HVRs generally comprise amino acid residues from the hypervariable loops and/or from the
"complementarity determining regions" (CDRs), the latter being of highest sequence variability and/or involved in antigen recognition. An HVR region as used herein comprise any number of residues located within positions 24-36 (for LI), 46-56 (for L2), 89-97 (for L3), 26-35B (for HI), 47-65 (for H2), and 93-102 (for H3). Therefore, an HVR includes residues in positions described previously:
A) 24-34 (LI), 50-52 (L2), 91-96 (L3), 26-32 (HI), 53-55 (H2), and 96-101 (H3) (Chothia and Lesk, J. Mol. Biol. 196:901-917 (1987);
B) 24-34 of LI, 50-56 of L2, 89-97 of L3, 31-35B of HI, 50-65 of H2, and 95-102 of H3 (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991).
C) 30-36 (LI), 46-55 (L2), 89-96 (L3), 30-35 (HI), 47-58 (H2), 93-100a-j (H3) (MacCallum et al. J. Mol. Biol. 262:732-745 (1996).
[0049] With the exception of CDR1 in VH, CDRs generally comprise the amino acid residues that form the hypervariable loops. CDRs also comprise "specificity determining residues," or "SDRs," which are residues that contact antigen. SDRs are contained within regions of the CDRs called abbreviated-CDRs, or a-CDRs. Exemplary a-CDRs (a-CDR-Ll, a-CDR-L2, a-CDR-L3, a-CDR-Hl, a-CDR-H2, and a-CDR-H3) occur at amino acid residues 31-34 of LI, 50-55 of L2, 89-96 of L3, 31-35B of HI, 50-58 of H2, and 95-102 of H3. (See Almagro and Fransson, Front. Biosci. 13: 1619-1633 (2008).) Unless otherwise indicated, HVR residues and other residues in the variable domain (e.g., FR residues) are numbered herein according to Kabat et al., supra.
[0050] An "immunoconjugate" is an antibody conjugated to one or more heterologous molecule(s), including but not limited to a cytotoxic agent.
[0051] An "individual" or "subject" is a mammal. Mammals include, but are not limited to, domesticated animals (e.g., cows, sheep, cats, dogs, and horses), primates (e.g., humans and non-human primates such as monkeys), rabbits, and rodents (e.g., mice and rats). In certain embodiments, the individual or subject is a human.
[0052] An "isolated" antibody is one which has been separated from a component of its natural environment. In some embodiments, an antibody is purified to greater than 95% or 99% purity as determined by, for example, electrophoretic (e.g., SDS-PAGE, isoelectric focusing (IEF), capillary electrophoresis) or chromatographic (e.g., ion exchange or reverse phase HPLC). For review of methods for assessment of antibody purity, see, e.g., Flatman et al., /. Chromatogr. B 848:79-87 (2007).
[0053] An "isolated" nucleic acid refers to a nucleic acid molecule that has been separated from a component of its natural environment. An isolated nucleic acid includes a nucleic acid molecule contained in cells that ordinarily contain the nucleic acid molecule, but the nucleic acid molecule is present extrachromosomally or at a chromosomal location that is different from its natural chromosomal location.
[0054] "Isolated nucleic acid encoding an anti-HCV antibody" refers to one or more nucleic acid molecules encoding antibody heavy and light chains (or fragments thereof), including such nucleic acid molecule(s) in a single vector or separate vectors, and such nucleic acid molecule(s) present at one or more locations in a host cell.
[0055] The term "monoclonal antibody" as used herein refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies
comprising the population are identical and/or bind the same epitope, except for possible variant antibodies, e.g., containing naturally occurring mutations or arising during production of a monoclonal antibody preparation, such variants generally being present in minor amounts. In contrast to polyclonal antibody preparations, which typically include different antibodies directed against different determinants (epitopes), each monoclonal antibody of a monoclonal antibody preparation is directed against a single determinant on an antigen. Thus, the modifier "monoclonal" indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method. For example, the monoclonal antibodies to be used in accordance with the present invention may be made by a variety of techniques, including but not limited to the hybridoma method, recombinant DNA methods, phage-display methods, and methods utilizing transgenic animals containing all or part of the human immunoglobulin loci, such methods and other exemplary methods for making monoclonal antibodies being described herein.
[0056] A "naked antibody" refers to an antibody that is not conjugated to a heterologous moiety (e.g., a cytotoxic moiety) or radiolabel. The naked antibody may be present in a pharmaceutical formulation.
[0057] "Native antibodies" refer to naturally occurring immunoglobulin molecules with varying structures. For example, native IgG antibodies are heterotetrameric glycoproteins of about 150,000 daltons, composed of two identical light chains and two identical heavy chains that are disulfide-bonded. From N- to C-terminus, each heavy chain has a variable region (VH), also called a variable heavy domain or a heavy chain variable domain, followed by three constant domains (CHI, CH2, and CH3). Similarly, from N- to C-terminus, each light chain has a variable region (VL), also called a variable light domain or a light chain variable domain, followed by a constant light (CL) domain. The light chain of an antibody may be assigned to one of two types, called kappa (κ) and lambda (λ), based on the amino acid sequence of its constant domain.
[0058] The term "package insert" is used to refer to instructions customarily included in commercial packages of therapeutic products, that contain information about the indications, usage, dosage, administration, combination therapy, contraindications and/or warnings concerning the use of such therapeutic products.
[0059] "Percent ( ) amino acid sequence identity" with respect to a reference polypeptide sequence is defined as the percentage of amino acid residues in a candidate sequence that are identical with the amino acid residues in the reference polypeptide sequence, after aligning
the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity. Alignment for purposes of determining percent amino acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or Megalign (DNASTAR) software. Those skilled in the art can determine appropriate parameters for aligning sequences, including any algorithms needed to achieve maximal alignment over the full length of the sequences being compared. For purposes herein, however, % amino acid sequence identity values are generated using the sequence comparison computer program ALIGN-2. The ALIGN-2 sequence comparison computer program was authored by Genentech, Inc., and the source code has been filed with user documentation in the U.S. Copyright Office,
Washington D.C., 20559, where it is registered under U.S. Copyright Registration No.
TXU510087. The ALIGN-2 program is publicly available from Genentech, Inc., South San Francisco, California, or may be compiled from the source code. The ALIGN-2 program should be compiled for use on a UNIX operating system, including digital UNIX V4.0D. All sequence comparison parameters are set by the ALIGN-2 program and do not vary.
[0060] In situations where ALIGN-2 is employed for amino acid sequence comparisons, the % amino acid sequence identity of a given amino acid sequence A to, with, or against a given amino acid sequence B (which can alternatively be phrased as a given amino acid sequence A that has or comprises a certain % amino acid sequence identity to, with, or against a given amino acid sequence B) is calculated as follows:
100 times the fraction X/Y
where X is the number of amino acid residues scored as identical matches by the sequence alignment program ALIGN-2 in that program's alignment of A and B, and where Y is the total number of amino acid residues in B. It will be appreciated that where the length of amino acid sequence A is not equal to the length of amino acid sequence B, the % amino acid sequence identity of A to B will not equal the % amino acid sequence identity of B to A. Unless specifically stated otherwise, all % amino acid sequence identity values used herein are obtained as described in the immediately preceding paragraph using the ALIGN-2 computer program.
[0061] The term "pharmaceutical formulation" refers to a preparation which is in such form as to permit the biological activity of an active ingredient contained therein to be effective, and which contains no additional components which are unacceptably toxic to a subject to which the formulation would be administered.
[0062] A "pharmaceutically acceptable carrier" refers to an ingredient in a pharmaceutical formulation, other than an active ingredient, which is nontoxic to a subject., A
pharmaceutically acceptable carrier includes, but is not limited to, a buffer, excipient, stabilizer, or preservative.
[0063] The term "HCV E2 protein," as used herein, refers to any native HCV E2 protein isolated from or identified in any vertebrate source, including mammals such as primates (e.g. humans and chimpanzees) and rodents (e.g., mice and rats) unless otherwise indicated. The term encompasses "full-length," unprocessed HCV E2 protein as well as any form of HCV E2 protein that results from processing in the cell or processing outside of the cell. The term also encompasses naturally occurring variants of HCV E2 protein, e.g., genotype variants or quasispecies. Exemplary naturally occurring variants of HCV E2 proteins can be found in, but not limited to, Simmonds P., J Gen Virol., (2004)., 85 (Pt 11):3173-88.
[0064] As used herein, the term "treatment" (and grammatical variations thereof such as "treat" or "treating") refers to clinical intervention designed to alter the natural course of the individual or cell being treated during the course of clinical pathology. Desirable effects of treatment include, but are not limited to, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, decreasing the rate of disease
progression, amelioration or palliation of the disease state, and remission or improved prognosis. In some embodiments, antibodies of the invention are used to delay development of a disease or to slow the progression of a disease. In some embodiments, the disease is an HCV-associated disease. In some embodiments, the HCV-associated disease is HCV infection. An individual is successfully "treated", for example, if one or more symptoms associated with HCV infection are mitigated or eliminated.
[0065] As used herein, the term "prevention" includes providing prophylaxis with respect to occurrence or recurrence of a disease in an individual. An individual may be predisposed to, susceptible to an HCV-associated disorder, or at risk of developing an HCV-associated disorder, but has not yet been diagnosed with the disorder. In some embodiments, an HCV- associated disorder is HCV infection. In some embodiments, an HCV-associated disorder is hepatocellular carcinoma.
[0066] The term "variable region" or "variable domain" refers to the domain of an antibody heavy or light chain that is involved in binding the antibody to antigen. The variable domains of the heavy chain and light chain (VH and VL, respectively) of a native antibody generally have similar structures, with each domain comprising four conserved framework regions (FRs) and three hypervariable regions (HVRs). (See, e.g., Kindt et al. Kuby Immunology, 6th
ed., W.H. Freeman and Co., page 91 (2007).) A single VH or VL domain may be sufficient to confer antigen-binding specificity. Furthermore, antibodies that bind a particular antigen may be isolated using a VH or VL domain from an antibody that binds the antigen to screen a library of complementary VL or VH domains, respectively. See, e.g., Portolano et al., J. Immunol. 150:880-887 (1993); Clarkson et al., Nature 352:624-628 (1991).
[0067] The term "vector," as used herein, refers to a nucleic acid molecule capable of propagating another nucleic acid to which it is linked. The term includes the vector as a self- replicating nucleic acid structure as well as the vector incorporated into the genome of a host cell into which it has been introduced. Certain vectors are capable of directing the expression of nucleic acids to which they are operatively linked. Such vectors are referred to herein as "expression vectors."
[0068] The term "about" as used herein refers to the usual error range for the respective value readily known to the skilled person in this technical field. Reference to "about" a value or parameter herein includes (and describes) embodiments that are directed to that value or parameter per se.
[0069] As used herein and in the appended claims, the singular forms "a," "an," and "the" include plural reference unless the context clearly indicates otherwise. For example, reference to an "antibody" is a reference to from one to many antibodies, such as molar amounts, and includes equivalents thereof known to those skilled in the art, and so forth.
[0070] It is understood that aspect and embodiments of the invention described herein include "comprising," "consisting," and "consisting essentially of aspects and embodiments.
III. Compositions and Methods
[0071] In one aspect, the invention provides methods for inhibiting, treating or preventing hepatitis C virus (HCV) infection in an individual comprising administering to the individual an effective amount of an anti-HCV antibody described herein. In some embodiments, an effective amount of an anti-HCV antibody is administered to an individual for inhibiting, treating or preventing HCV cellular entry in an individual. In some embodiments, an effective amount of an anti-HCV antibody is administered to an individual for inhibiting, treating or preventing HCV spread in an individual. In some embodiments, an effective amount of an anti-HCV antibody is administered to an individual for inhibiting, treating or preventing an HCV-associated disease in the individual. In some embodiments herein, the HCV is a drug-resistant HCV.
[0072] With respect to all methods described herein, reference to an anti-HCV antibody also includes compositions comprising one or more of those agents. Such compositions may further comprise suitable excipients, such as pharmaceutically acceptable excipients (carriers) including buffers, acids, bases, sugars, diluents, preservatives, and the like, which are well known in the art and are described herein. The present methods can be used alone or in combination with other conventional methods of treatment (e.g., antivirals).
A. Anti-HCV antibodies
[0073] The methods of the present invention use an anti-HCV antibody, which term refers to an anti-HCV antibody that binds to HCV E2 protein. In some embodiments, the HCV E2 protein is expressed on a hepatitis C viral surface and therefore inhibits HCV cellular entry by preventing binding of HCV E2 protein to a receptor expressed on a host cell surface (such as CD81 expressed on a human cell surface). See Figure 1. The anti-HCV antibodies described herein may have one or more of the following characteristics: (a) bind HCV E2 protein or variants thereof (such as HCV E2 glycosylation variants); (b) block binding of HCV E2 protein to a host cell (such as a human host cell); (c) inhibit HCV entry into a host cell; (d) inhibit and/ or prevent HCV infection of a host cell (such as a host cell in an individual); (e) inhibit HCV spread in an individual (such as a human); (f) treat and/or prevent emergence of drug-resistant HCV; (g) enhance inhibition of HCV infection and/or spread by other antivirals (such as HCV protease inhibitors); and (h) treat and/or prevent an HCV-associated disease (such as hepatocellular carcinoma). The activities of anti-HCV antibodies may be measured in vitro and/or in vivo.
[0074] In one aspect, the invention provides an anti-HCV antibody comprising at least one, two, three, four, five, or six hypervariable regions (HVRs) selected from (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: l; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:2; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO:3; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO:4; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
[0075] In one aspect, the invention provides an anti-HCV antibody comprising at least one, at least two, or all three VH HVR sequences selected from (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: l; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:2; and (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO:3. In one embodiment, the anti-HCV antibody comprises HVR-H3 comprising the amino acid sequence of SEQ ID NO:3. In another embodiment, the anti-HCV antibody comprises HVR-
H3 comprising the amino acid sequence of SEQ ID NO:3 and HVR-L3 comprising the amino acid sequence of SEQ ID NO:6. In a further embodiment, the anti-HCV antibody comprises HVR-H3 comprising the amino acid sequence of SEQ ID NO:3, HVR-L3 comprising the amino acid sequence of SEQ ID NO:6, and HVR-H2 comprising the amino acid sequence of SEQ ID NO:2. In a further embodiment, the anti-HCV antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: l ; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:2; and (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO:3.
[0076] In another aspect, the invention provides an anti-HCV antibody comprising at least one, at least two, or all three VH HVR sequences selected from (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:4; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (c) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6. In one embodiment, the anti-HCV antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:4; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (c) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
[0077] In another aspect, the anti-HCV antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: l ; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:2; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO:3; (d) HVR- Ll comprising the amino acid sequence of SEQ ID NO:4; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising an amino acid sequence selected from SEQ ID NO:6.
[0078] As described herein, SEQ ID NO: 1 refers to amino acid sequence
GXiSX2TSGYWN, wherein Χ is D or E, and X2 is I or L. SEQ ID NO:2 refers to amino acid sequence YISYSGSTYYXiX2SLRS, wherein Χ is N or S, and wherein X2 is P or L. SEQ ID NO:3 refers to amino acid sequence ALITTXiTYAMDY, wherein Xl is S or T. SEQ ID NO:4 refers to amino acid sequence RASES VX1GYGX2SFLH, wherein Xl is D or S, and X2 is N or Y. SEQ ID NO:5 refers to amino acid sequence LASNLNS. SEQ ID NO:6 refers to amino acid sequence QQNNVDPWT.
[0079] In certain embodiments, the anti-HCV antibody is not an antibody comprising (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 11 ; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 15; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising an amino acid sequence selected from SEQ ID NO:6.
[0080] In another aspect, an anti-HCV antibody of the invention comprises a heavy chain variable region and a light chain variable region,
(a) wherein the heavy chain variable region comprises three HVR sequences:
(i) HVR-Hl comprising GXiSX2TSGYWN (SEQ ID NO: l), wherein Xi is D or E, and X2 is I or L;
(ii) HVR-H2 comprising YISYSGSTYYXiX2SLRS (SEQ ID NO:2), wherein Xl is N or S, and wherein X2 is P or L; and
(iii) HVR-H3 comprising ALITTXiTYAMDY (SEQ ID NO:3), wherein Xi is S or T; and/or
(b) wherein the light chain variable region domain comprises three HVR sequences:
(i) HVR-Ll comprising RASES VXiGYGX2SFLH (SEQ ID NO:4), wherein Xi is D or S, and X2 is N or Y;
(ii) HVR-L2 comprising LASNLNS (SEQ ID NO:5); and
(iii) HVR-L3 comprising QQNNVDPWT (SEQ ID NO:6).
[0081] In another aspect, an anti-HCV antibody of the invention comprises a heavy chain variable region and a light chain variable region,
(a) wherein the heavy chain variable region comprises three HVR sequences:
(i) HVR-Hl comprising GDSITSGYWN (SEQ ID NO:7) or GESLTSGYWN (SEQ ID NO:8);
(ii) HVR-H2 comprising YISYSGSTYYNPSLRS (SEQ ID NO:9),
YISYSGSTYYSLSLRS (SEQ ID NO: 10), or YISYSGSTYYNLSLRS (SEQ ID NO: 1 1); and
(iii) HVR-H3 comprising ALITTSTYAMDY (SEQ ID NO: 12) or
ALITTTTYAMDY (SEQ ID NO: 13); and/or
(b) wherein the light chain variable region domain comprises three HVR sequences:
(i) HVR-Ll comprising RASES VXiGYGX2SFLH (SEQ ID NO:4), wherein Xl is D or S, and X2 is N or Y;
(ii) HVR-L2 comprising LASNLNS (SEQ ID NO:5); and
(iii) HVR-L3 comprising QQNNVDPWT (SEQ ID NO:6)
[0082] In another aspect, an anti-HCV antibody of the invention comprises a heavy chain variable region and a light chain variable region,
(a) wherein the heavy chain variable region comprises three HVR sequences:
(i) HVR-Hl comprising GXiSX2TSGYWN (SEQ ID NO: l), wherein Xl is D or E, and X2 is I or L;
(ii) HVR-H2 comprising YISYSGSTYYXiX2SLRS (SEQ ID NO:2), wherein Xi is N or S, and wherein X2 is P or L; and
(iii) HVR-H3 comprising ALITTX \ TY AMD Y (SEQ ID NO:3), wherein Χ is S or T; and/or
(b) wherein the light chain variable region domain comprises three HVR sequences:
(i) HVR-L1 comprising RASES VDGYGYSFLH (SEQ ID NO: 14),
RASES VDGYGNSFLH (SEQ ID NO: 15), or RASESVSGYGYSFLH (SEQ ID NO: 16)
(ii) HVR-L2 comprising LASNLNS (SEQ ID NO:5); and
(iii) HVR-L3 comprising QQNNVDPWT (SEQ ID NO:6)
[0083] In another aspect, the invention provides an anti-HCV antibody comprising (a) HVR-Hl comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NOs:9 or 10; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 12; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 14; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
[0084] In another aspect, the invention provides an anti-HCV antibody comprising (a) HVR-Hl comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NOs:9 or 10; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 15; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
[0085] In another aspect, the invention provides an anti-HCV antibody comprising (a) HVR-Hl comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 11 ; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 12; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NOs: 14 or 15; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
[0086] In another aspect, the invention provides an anti-HCV antibody comprising (a) HVR-Hl comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 11 ; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 14; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
[0087] In another aspect, the invention provides an anti-HCV antibody comprising (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 8; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:9; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 16; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
[0088] In another aspect, an anti-HCV antibody of the invention comprises a heavy chain variable region sequence of
EVQLQESGPGLVKPSETLSLTCTVSGDSITSGYWNWIRQPPGRALEWMGYISYSGST YYNPSLRSRITISRDTSKNQYSLRLSSVTAADTAMYYCALITTSTYAMDYWGQGTTV TVSS (SEQ ID NO: 17),
EVQLQESGPGLVKPSETLSLTCTVSGDSITSGYWNWIRQPPGRALEWMGYISYSGST YYSLSLRSRITISRDTSKNQYSLRLSSVTAADTAMYYCALITTSTYAMDYWGQGTTV TVSS (SEQ ID NO: 18),
EVQLQESGPGLVKPSETLSLTCTVSGDSITSGYWNWIRQPPGRALEWMGYISYSGST YYSLSLRSRITISRDTSKNQYSLRLSSVTAADTAMYYCALITTTTYAMDYWGQGTTV TVSS (SEQ ID NO: 19),
EVQLQESGPGLVKPSETLSLTCTVSGDSITSGYWNWIRQPPGRALEWMGYISYSGST YYNPSLRSRITISRDTSKNQYSLRLSSVTAADTAMYYCALITTTTYAMDYWGQGTTV TVSS (SEQ ID NO: 20),
EVQLQESGPGLVKPSETLSLTCTVSGDSITSGYWNWIRQPPGRALEWMGYISYSGST YYNLSLRSRITISRDTSKNQYSLRLSSVTAADTAMYYCALITTTTYAMDYWGQGTTV TVSS (SEQ ID NO:21)
EVQLQESGPGLVKPSETLSLTCTVSGDSITSGYWNWIRQPPGRALEWMGYISYSGST YYNLSLRSRITISRDTSKNQYSLRLSSVTAADTAMYYCALITTSTYAMDYWGQGTTV TVSS (SEQ ID NO:22), or
EVQLQESGPGLVKPSETLSLTCTVSGESLTSGYWNWIRQPPGRALEWMGYISYSGST
YYNPSLRSRITISRDTSKNQYSLRLSSVTAADTAMYYCALITTTTYAMDYWGQGTTV
TVSS (SEQ ID NO:23); and/or
a light chain variable region sequence of
DIVLTQSPSSLSASVGDRVTITCRASESVDGYGYSFLHWFQQKPGKAPKLLIYLASNL NSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQNNVDPWTFGQGTKLEIK (SEQ ID NO:24),
DIVLTQSPSSLSASVGDRVTITCRASESVDGYGNSFLHWFQQKPGKAPKLLIYLASNL NSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQNNVDPWTFGQGTKLEIK (SEQ ID NO:25), or
DIVLTQSPSSLSASVGDRVTITCRASESVSGYGYSFLHWFQQKPGKAPKLLIYLASNL NSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQNNVDPWTFGQGTKLEIK (SEQ ID NO:26).
[0089] In certain embodiments, an anti-HCV antibody of the invention is not an antibody comprising a heavy chain variable region sequence of
EVQLQESGPGLVKPSETLSLTCTVSGDSITSGYWNWIRQPPGRALEWMGYISYSGST
YYNLSLRSRITISRDTSKNQYSLRLSSVTAADTAMYYCALITTTTYAMDYWGQGTTV
TVSS (SEQ ID NO:21), and
a light chain variable region sequence of
DIVLTQSPSSLSASVGDRVTITCRASESVDGYGNSFLHWFQQKPGKAPKLLIYLASNL NSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQNNVDPWTFGQGTKLEIK (SEQ ID NO:25).
[0090] In another aspect, an anti-HCV antibody of the invention comprises a heavy chain sequence of
EVQLQESGPGLVKPSETLSLTCTVSGDSITSGYWNWIRQPPGRALEWMGYISYSGST
YYNPSLRSRITISRDTSKNQYSLRLSSVTAADTAMYYCALITTSTYAMDYWGQGTTV
TVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFP
AVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPC
PAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHN
AKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQ
PREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLD
SDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG (SEQ ID
NO:27), or
EVQLQESGPGLVKPSETLSLTCTVSGDSITSGYWNWIRQPPGRALEWMGYISYSGST
YYSLSLRSRmSRDTSKNQYSLRLSSVTAADTAMYYCALITTSTYAMDYWGQGTTV
TVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFP
AVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPC
PAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHN
AKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQ
PREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLD
SDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG (SEQ ID
NO:28); and/or
the light chain sequence of
DIVLTQSPSSLSASVGDRVTITCRASESVDGYGYSFLHWFQQKPGKAPKLLIYLASNL NSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQNNVDPWTFGQGTKLEIKRTVAA PSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSK DSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO:30).
[0091] The invention also provides anti-HCV antibodies comprising three CDRs of the heavy chain and three CDRs of the light chain of anti-HCV antibodies described herein (e.g., V361, V362, VI, V6, V79, V317, V335, V355). The invention also provides anti-HCV antibodies comprising a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence SGYWN (SEQ ID NO:33), CDR H2 comprising the amino acid sequence of SEQ ID NO:9, 10, or 11, CDR H3 comprising the amino acid sequence ITTSTYAMDY (SEQ ID NO:34) or ITTTTYAMDY (SEQ ID NO:35); and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, 15, or 16, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the anti-HCV antibody is not an antibody comprising a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 10, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33,
CDR H2 comprising the amino acid sequence of SEQ ID NO:9, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:34; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO:9, CDR H3 comprising the amino acid sequence of SEQ ID NO:35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 16, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6. In some embodiments, the anti-HCV antibody comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO: 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:34; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 15, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
[0092] In some of the above embodiments, an anti-HCV antibody is humanized. In one embodiment, an anti-HCV antibody comprises HVRs as in any of the above embodiments, and further comprises an acceptor human framework, e.g. a human immunoglobulin framework or a human consensus framework. In another embodiment, an anti-HCV antibody comprises HVRs as in any of the above embodiments, and further comprises a VL comprising an FR1, FR2, FR3, or FR4 sequence as shown in Figure 2A. In another embodiment, an anti-HCV antibody comprises HVRs as in any of the above embodiments, and further comprises a VH comprising an FR1, FR2, FR3, or FR4 sequence as shown in Figure 2B.
[0093] In another aspect, an anti-HCV antibody comprises a heavy chain variable domain (VH) sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to the amino acid sequence of SEQ ID NOs: 17, 18, 19, 20, 21, 22, or 23. In certain embodiments, a VH sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity contains substitutions (e.g., conservative
substitutions), insertions, or deletions relative to the reference sequence, but an anti-HCV antibody comprising that sequence retains the ability to bind to HCV E2 protein. In certain embodiments, a total of 1 to 10 amino acids have been substituted, inserted and/or deleted in SEQ ID NOs: 17, 18, 19, 20, 21, 22, or 23. In certain embodiments, substitutions, insertions, or deletions occur in regions outside the HVRs (i.e., in the FRs). Optionally, the anti-HCV antibody comprises the VH sequence in SEQ ID NOs: 17, 18, 19, 20, 21, 22, or 23, including post-translational modifications of that sequence. In a particular embodiment, the VH comprises one, two or three HVRs selected from: (a) HVR-H1 comprising the amino acid sequence of SEQ ID NOs:7 or 8, (b) HVR-H2 comprising the amino acid sequence of SEQ ID NOs:9, 10, or 11, and (c) HVR-H3 comprising the amino acid sequence of SEQ ID NOs: 12 or 13. In certain embodiments, the VH is not a VH comprising three HVRs consisting of: (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:7, (b) HVR- H2 comprising the amino acid sequence of SEQ ID NO: 11, and (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13.
[0094] In another aspect, an anti-HCV antibody is provided, wherein the antibody comprises a light chain variable domain (VL) having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to the amino acid sequence of SEQ ID NOs:24, 25, or 26. In certain embodiments, a VL sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity contains substitutions (e.g., conservative substitutions), insertions, or deletions relative to the reference sequence, but an
anti-HCV antibody comprising that sequence retains the ability to bind to HCV E2 protein. In certain embodiments, a total of 1 to 10 amino acids have been substituted, inserted and/or deleted in SEQ ID NOs:24, 25, or 26. In certain embodiments, the substitutions, insertions, or deletions occur in regions outside the HVRs (i.e., in the FRs). Optionally, the anti-HCV antibody comprises the VL sequence in SEQ ID NOs:24, 25, or 26, including post- translational modifications of that sequence. In a particular embodiment, the VL comprises one, two or three HVRs selected from (a) HVR-L1 comprising the amino acid sequence of SEQ ID NOs: 14, 15, or 16; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (c) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6. In certain embodiments, the VL is not a VL comprising three HVRs consisting of: (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 15; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (c) HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
[0095] In another aspect, an anti-HCV antibody is provided, wherein the antibody comprises a VH as in any of the embodiments provided above, and a VL as in any of the embodiments provided above. In one embodiment, the antibody comprises the VH and VL sequences of SEQ ID NO: 17 and SEQ ID NO:24, respectively, including post-translational modifications of those sequences. In another embodiment, the antibody comprises the VH and VL sequences of SEQ ID NO: 18 and SEQ ID NO:24, respectively, including post- translational modifications of those sequences. In yet another embodiment, the antibody comprises the VH and VL sequences of SEQ ID NO: 19 and SEQ ID NO:25, respectively, including post-translational modifications of those sequences. In still another embodiment, the antibody comprises the VH and VL sequences of SEQ ID NO:20 and SEQ ID NO:25, respectively, including post-translational modifications of those sequences. In another embodiment, the antibody comprises the VH and VL sequences of SEQ ID NO:21 and SEQ ID NO:24, respectively, including post-translational modifications of those sequences. In another embodiment, the antibody comprises the VH and VL sequences of SEQ ID NO:22 and SEQ ID NO:24, respectively, including post-translational modifications of those sequences. In yet another embodiment, the antibody comprises the VH and VL sequences of SEQ ID NO:23 and SEQ ID NO:26, respectively, including post-translational modifications of those sequences. In another embodiment, the antibody comprises the VH and VL sequences of SEQ ID NO:22 and SEQ ID NO:25, respectively, including post-translational modifications of those sequences.
[0096] In a further aspect, the invention provides an antibody that binds to the same epitope as an anti-HCV antibody provided herein. For example, in certain embodiments, an anti- HCV antibody is provided that binds to the same epitope as an anti-HCV antibody
comprising a VH sequence of SEQ ID NO: 17 and a VL sequence of SEQ ID NO:24. In certain embodiments, an anti-HCV antibody is provided that binds to an epitope within a fragment of HCV E2 protein consisting of the amino acid sequence of QLINTNGSWHIN (SEQ ID NO:32). In certain embodiments, an anti-HCV antibody is provided that binds to an epitope within a fragment of HCV E2 protein consisting of the amino acid sequence of QLINTNGSWHIN (SEQ ID NO:32) or a homologous amino acid sequence isolated from an HCV genotype selected from the group consisting of genotype 1 (e.g., genotype la and genotype lb), genotype 2 (e.g. , genotype 2a, genotype 2b, genotype 2c), genotype 3 (e.g., genotype 3a), genotype 4 (e.g., genotype 4a), genotype 5, and genotype 6. In certain embodiments, an anti-HCV antibody is provided that binds to an epitope within a fragment of an HCV E2 protein consisting of an amino acid sequence similar to the sequence of amino acids 412 to 423 of an HCV E2 protein isolated from HCV genotype 1. As used in these embodiments, the term "homologous amino acid sequence" refers to an epitope that has a similar amino acid sequence to the amino acid sequence of QLINTNGSWHIN (SEQ ID NO:32) found in genotype la or genotype lb HCV E2 protein. For example, one or more amino acids may be substituted in another HCV genotye as compared to the sequence of QLINTNGSWHIN (SEQ ID NO:32). In some embodiments herein, the HCV E2 protein is soluble HCV E2 protein. In some embodiments herein, the HCV E2 protein is an HCV E2 protein fragment (such as amino acids 1 to 661 of HCV E2 protein isolated from HCV genotype la). Exemplary HCV E2 proteins can be found in, but not limited to, Bukh J et al., Semin Liv Dis., (1995)., 15(l):41-63.
[0097] In a further aspect of the invention, an anti-HCV antibody according to any of the above embodiments is a monoclonal antibody, including a chimeric, humanized or human antibody. In one embodiment, an anti-HCV antibody is an antibody fragment, e.g., a Fv, Fab, Fab'-SH, scFv, diabody, or F(ab')2 fragment. In another embodiment, the antibody is a full length antibody, e.g., an intact IgGl antibody or other antibody class or isotype as defined herein.
[0098] In a further aspect, an anti-HCV antibody according to any of the above
embodiments may incorporate any of the features, singly or in combination, as described in Sections 1-7 below:
1. Antibody Affinity
[0099] In certain embodiments, an anti-HCV antibody provided herein has a dissociation constant (Kd) of < ΙμΜ, < 100 nM, < 10 nM, < 1 nM, < 0.1 nM, < 0.01 nM, or < 0.001 nM (e.g. 10~8 M or less, e.g. from 10~8 M to 10"13 M, e.g., from 10"9 M to 10"13 M). In some embodiments herein, an anti-HCV antibody provided herein has a Kd for a binding partner (such as HCV E2 protein or fragment thereof) of less than about any of about 1.0 mM, 500 μΜ, 100 μΜ, 50μΜ, 25 μΜ, 10 μΜ, 5 μΜ, 1 μΜ, 900 ηΜ, 800 ηΜ, 700 ηΜ, 600 ηΜ, 500 ηΜ, 400 ηΜ, 350 ηΜ, 300 ηΜ, 250 ηΜ, 200 ηΜ, 150 ηΜ, 100 ηΜ,95 ηΜ, 90 ηΜ, 85 ηΜ, 80 ηΜ, 75 ηΜ, 70 ηΜ, 65 ηΜ, 60 ηΜ, 55 ηΜ, 50 ηΜ, 45 ηΜ, 40 ηΜ, 35 ηΜ, 30 ηΜ, 25 ηΜ, 20 ηΜ, 15 ηΜ, 10 ηΜ, 5 nM, 1 ηΜ, 900 ρΜ, 800 ρΜ, 700 ρΜ, 600 ρΜ, 500 ρΜ, 400 ρΜ, 300 ρΜ, 200 ρΜ, 100 ρΜ, 50 ρΜ, 25 ρΜ, 12.5 ρΜ, 6.25 ρΜ, 5 ρΜ, 4 ρΜ, or 3ρΜ, inclusive, including any values in between these numbers. In some embodiments, an anti-HCV antibody described herein binds to HCV E2 protein or fragment thereof with a higher affinity compared to the binding of a parent antibody (such as MRCT10.1) to HCV E2 protein or fragment thereof. In some aspects, and anti-HCV antibody binds to HCV E2 protein or a fragment thereof with at least any of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 ,17, 18, 19, or 20, inclusive, including any value in between these numbers, higher fold affinity (or lowed KD) compared to the binding of the parent antibody (such as MRCT10.1) to the HCV E2 protein or fragment thereof. In some embodiments, an anti-HCV antibody described herein demonstrates a similar binding affinity to HCV E2 protein or fragment thereof as compared to the binding affinity of a parent antibody (such as MRCT10.1) to HCV E2 protein or fragment thereof. In further embodiments, the anti-HCV antibody has a higher HCV inhibitory potency (such as lower EC90) as compared to the HCV inhibitory potency of a parent antibody (such as MRCT10.1).
[0100] Methods for measuring Kd are well known in the art. For example, Kd is measured by a radiolabeled antigen binding assay (RIA) performed with the Fab version of an antibody of interest and its antigen as described by the following assay. Solution binding affinity of
Fabs for antigen is measured by equilibrating Fab with a minimal concentration of ( 125 I)- labeled antigen in the presence of a titration series of unlabeled antigen, then capturing bound antigen with an anti-Fab antibody-coated plate (see, e.g., Chen et al., J. Mol. Biol. 293:865- 881(1999)). To establish conditions for the assay, MICROTITER® multi-well plates
(Thermo Scientific) are coated overnight with 5 μg/ml of a capturing anti-Fab antibody (Cappel Labs) in 50 mM sodium carbonate (pH 9.6), and subsequently blocked with 2%
(w/v) bovine serum albumin in PBS for two to five hours at room temperature (approximately 23°C). In a non-adsorbent plate (Nunc #269620), 100 pM or 26 pM [125I]- antigen are mixed with serial dilutions of a Fab of interest (e.g., consistent with assessment of the anti-VEGF antibody, Fab- 12, in Presta et al., Cancer Res. 57:4593-4599 (1997)). The Fab of interest is then incubated overnight; however, the incubation may continue for a longer period (e.g., about 65 hours) to ensure that equilibrium is reached. Thereafter, the mixtures are transferred to the capture plate for incubation at room temperature (e.g., for one hour). The solution is then removed and the plate washed eight times with 0.1% polysorbate 20 (TWEEN-20®) in PBS. When the plates have dried, 150 μΐ/well of scintillant (MICROSCINT-20™; Packard) is added, and the plates are counted on a TOPCOUNT™ gamma counter (Packard) for ten minutes. Concentrations of each Fab that give less than or equal to 20% of maximal binding are chosen for use in competitive binding assays.
[0101] According to another embodiment, Kd is measured using surface plasmon resonance assays using a BIACORE®-2000 or a BIACORE®-3000 (BIAcore, Inc.,
Piscataway, NJ) at 25°C with immobilized antigen CM5 chips at -10 response units (RU). Briefly, carboxymethylated dextran biosensor chips (CM5, BIACORE, Inc.) are activated with N-ethyl-N'- (3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC) and N- hydroxysuccinimide (NHS) according to the supplier's instructions. Antigen is diluted with 10 mM sodium acetate, pH 4.8, to 5 μg/ml (-0.2 μΜ) before injection at a flow rate of 5 μΐ/minute to achieve approximately 10 response units (RU) of coupled protein. Following the injection of antigen, 1 M ethanolamine is injected to block unreacted groups. For kinetics measurements, two-fold serial dilutions of Fab (0.78 nM to 500 nM) are injected in PBS with 0.05% polysorbate 20 (TWEEN-20™) surfactant (PBST) at 25°C at a flow rate of approximately 25 μΐ/min. Association rates (kon) and dissociation rates (k0ff) are calculated using a simple one-to-one Langmuir binding model (BIACORE Evaluation Software version 3.2) by simultaneously fitting the association and dissociation sensorgrams. The equilibrium dissociation constant (Kd) is calculated as the ratio k0ff/kon See, e.g., Chen et al., J. Mol. Biol. 293:865-881 (1999). If the on-rate exceeds 106 M" 1 s" 1 by the surface plasmon resonance assay above, then the on-rate can be determined by using a fluorescent quenching technique that measures the increase or decrease in fluorescence emission intensity (excitation = 295 nm; emission = 340 nm, 16 nm band-pass) at 25°C of a 20 nM anti-antigen antibody (Fab form) in PBS, pH 7.2, in the presence of increasing concentrations of antigen as measured in a spectrometer, such as a stop-flow equipped spectrophometer
(Aviv Instruments) or a 8000-series SLM-AMINCO spectrophotometer
(ThermoSpectronic) with a stirred cuvette. In some embodiments, the Kd is measure as described in Example 2.
2. Antibody Fragments
[0102] In certain embodiments, an antibody provided herein is an antibody fragment. Antibody fragments include, but are not limited to, Fab, Fab', Fab'-SH, F(ab')2, Fv, and scFv fragments, and other fragments described below. For a review of certain antibody fragments, see Hudson et al. Nat. Med. 9: 129-134 (2003). For a review of scFv fragments, see, e.g., Pluckthiin, in The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., (Springer- Verlag, New York), pp. 269-315 (1994); see also WO 93/16185; and U.S. Patent Nos. 5,571,894 and 5,587,458. For discussion of Fab and F(ab')2 fragments comprising salvage receptor binding epitope residues and having increased in vivo half-life, see U.S. Patent No. 5,869,046.
[0103] Diabodies are antibody fragments with two antigen-binding sites that may be bivalent or bispecific. See, for example, EP 404,097; WO 1993/01161; Hudson et al., Nat. Med. 9: 129-134 (2003); and Hollinger et al., Proc. Natl. Acad. Sci. USA 90: 6444-6448 (1993). Triabodies and tetrabodies are also described in Hudson et al., Nat. Med. 9: 129-134 (2003).
[0104] Single-domain antibodies are antibody fragments comprising all or a portion of the heavy chain variable domain or all or a portion of the light chain variable domain of an antibody. In certain embodiments, a single-domain antibody is a human single-domain antibody (Domantis, Inc., Waltham, MA; see, e.g., U.S. Patent No. 6,248,516 Bl).
[0105] Antibody fragments can be made by various techniques, including but not limited to proteolytic digestion of an intact antibody as well as production by recombinant host cells (e.g. E. coli or phage), as described herein.
3. Chimeric and Humanized Antibodies
[0106] In certain embodiments, an antibody provided herein is a chimeric antibody.
Certain chimeric antibodies are described, e.g., in U.S. Patent No. 4,816,567; and Morrison et al., Proc. Natl. Acad. Sci. USA, 81:6851-6855 (1984)). In one example, a chimeric antibody comprises a non-human variable region (e.g., a variable region derived from a mouse, rat, hamster, rabbit, or non-human primate, such as a monkey) and a human constant region. In a further example, a chimeric antibody is a "class switched" antibody in which the class or
subclass has been changed from that of the parent antibody. Chimeric antibodies include antigen-binding fragments thereof.
[0107] In certain embodiments, a chimeric antibody is a humanized antibody. Typically, a non-human antibody is humanized to reduce immunogenicity to humans, while retaining the specificity and affinity of the parental non-human antibody. Generally, a humanized antibody comprises one or more variable domains in which HVRs, e.g., CDRs, (or portions thereof) are derived from a non-human antibody, and FRs (or portions thereof) are derived from human antibody sequences. A humanized antibody optionally will also comprise at least a portion of a human constant region. In some embodiments, some FR residues in a humanized antibody are substituted with corresponding residues from a non-human antibody (e.g., the antibody from which the HVR residues are derived), e.g., to restore or improve antibody specificity or affinity.
[0108] Humanized antibodies and methods of making them are reviewed, e.g., in Almagro and Fransson, Front. Biosci. 13: 1619-1633 (2008), and are further described, e.g., in
Riechmann et al., Nature 332:323-329 (1988); Queen et al., Proc. Nat'lAcad. Sci. USA 86: 10029-10033 (1989); US Patent Nos. 5, 821,337, 7,527,791, 6,982,321, and 7,087,409; Kashmiri et al., Methods 36:25-34 (2005) (describing SDR (a-CDR) grafting); Padlan, Mol. Immunol. 28:489-498 (1991) (describing "resurfacing"); Dall'Acqua et al., Methods 36:43-60 (2005) (describing "FR shuffling"); and Osbourn et al., Methods 36:61-68 (2005) and Klimka et al., Br. J. Cancer, 83:252-260 (2000) (describing the "guided selection" approach to FR shuffling).
[0109] Human framework regions that may be used for humanization include but are not limited to: framework regions selected using the "best-fit" method (see, e.g., Sims et al. J. Immunol. 151:2296 (1993)); framework regions derived from the consensus sequence of human antibodies of a particular subgroup of light or heavy chain variable regions (see, e.g., Carter et al. Proc. Natl. Acad. Sci. USA, 89:4285 (1992); and Presta et al. J. Immunol, 151:2623 (1993)); human mature (somatically mutated) framework regions or human germline framework regions (see, e.g., Almagro and Fransson, Front. Biosci. 13: 1619-1633 (2008)); and framework regions derived from screening FR libraries (see, e.g., Baca et al., J. Biol. Chem. 272: 10678-10684 (1997) and Rosok et al., J. Biol. Chem. 271:22611-22618 (1996)).
4. Human Antibodies
[0110] In certain embodiments, an antibody provided herein is a human antibody. Human antibodies can be produced using various techniques known in the art. Human antibodies are described generally in van Dijk and van de Winkel, Curr. Opin. Pharmacol. 5: 368-74 (2001) and Lonberg, Curr. Opin. Immunol. 20:450-459 (2008).
[0111] Human antibodies may be prepared by administering an immunogen to a transgenic animal that has been modified to produce intact human antibodies or intact antibodies with human variable regions in response to antigenic challenge. Such animals typically contain all or a portion of the human immunoglobulin loci, which replace the endogenous
immunoglobulin loci, or which are present extrachromosomally or integrated randomly into the animal's chromosomes. In such transgenic mice, the endogenous immunoglobulin loci have generally been inactivated. For review of methods for obtaining human antibodies from transgenic animals, see Lonberg, Nat. Biotech. 23: 1117-1125 (2005). See also, e.g., U.S. Patent Nos. 6,075,181 and 6,150,584 describing XENOMOUSE™ technology; U.S. Patent No. 5,770,429 describing HUMAB® technology; U.S. Patent No. 7,041,870 describing K-M MOUSE® technology, and U.S. Patent Application Publication No. US 2007/0061900, describing VELOCIMOUSE® technology). Human variable regions from intact antibodies generated by such animals may be further modified, e.g., by combining with a different human constant region.
[0112] Human antibodies can also be made by hybridoma-based methods. Human myeloma and mouse-human heteromyeloma cell lines for the production of human monoclonal antibodies have been described. (See, e.g., Kozbor J. Immunol., 133: 3001 (1984); Brodeur et al., Monoclonal Antibody Production Techniques and Applications, pp. 51-63 (Marcel Dekker, Inc., New York, 1987); and Boerner et al., J. Immunol., 147: 86 (1991).) Human antibodies generated via human B-cell hybridoma technology are also described in Li et al, Proc. Nad. Acad. Sti. USA, 103:3557-3562 (2006). Additional methods include those described, for example, in U.S. Patent No. 7,189,826 (describing production of monoclonal human IgM antibodies from hybridoma cell lines) and Ni, Xiandai Mianyixue, 26(4):265-268 (2006) (describing human-human hybridomas). Human hybridoma technology (Trioma technology) is also described in Vollmers and Brandlein, Histology and Histopathology, 20(3):927-937 (2005) and Vollmers and Brandlein, Methods and Findings in Experimental and Clinical Pharmacology, 27(3): 185-91 (2005).
[0113] Human antibodies may also be generated by isolating Fv clone variable domain sequences selected from human-derived phage display libraries. Such variable domain sequences may then be combined with a desired human constant domain. In some
embodiments, an anti-HCV antibody is generated using the method described in Example 1. Techniques for selecting human antibodies from antibody libraries are described below.
5. Library-Derived Antibodies
[0114] Antibodies of the invention may be isolated by screening combinatorial libraries for antibodies with the desired activity or activities. For example, a variety of methods are known in the art for generating phage display libraries and screening such libraries for antibodies possessing the desired binding characteristics. Such methods are reviewed, e.g., in Hoogenboom et al. in Methods in Molecular Biology 178: 1-37 (O'Brien et al., ed., Human Press, Totowa, NJ, 2001) and further described, e.g., in the McCafferty et al., Nature
348:552-554; Clackson et al., Nature 352: 624-628 (1991); Marks et al., /. Mol. Biol. 222: 581-597 (1992); Marks and Bradbury, in Methods in Molecular Biology 248: 161-175 (Lo, ed., Human Press, Totowa, NJ, 2003); Sidhu et al., /. Mol. Biol. 338(2): 299-310 (2004); Lee et al., /. Mol. Biol. 340(5): 1073-1093 (2004); Fellouse, Proc. Natl. Acad. Sci. USA 101(34): 12467-12472 (2004); and Lee et al., /. Immunol. Methods 284(1-2): 119-132(2004).
[0115] In certain phage display methods, repertoires of VH and VL genes are separately cloned by polymerase chain reaction (PCR) and recombined randomly in phage libraries, which can then be screened for antigen-binding phage as described in Winter et al., Ann. Rev. Immunol., 12: 433-455 (1994). Phage typically display antibody fragments, either as single- chain Fv (scFv) fragments or as Fab fragments. Libraries from immunized sources provide high-affinity antibodies to the immunogen without the requirement of constructing hybridomas. Alternatively, the naive repertoire can be cloned (e.g., from human) to provide a single source of antibodies to a wide range of non-self and also self-antigens without any immunization as described by Griffiths et al., EMBO J, 12: 725-734 (1993). Finally, naive libraries can also be made synthetically by cloning unrearranged V-gene segments from stem cells, and using PCR primers containing random sequence to encode the highly variable CDR3 regions and to accomplish rearrangement in vitro, as described by Hoogenboom and Winter, /. Mol. Biol., 227: 381-388 (1992). Patent publications describing human antibody phage libraries include, for example: US Patent No. 5,750,373, and US Patent Publication Nos. 2005/0079574, 2005/0119455, 2005/0266000, 2007/0117126, 2007/0160598,
2007/0237764, 2007/0292936, and 2009/0002360.
[0116] Antibodies or antibody fragments isolated from human antibody libraries are considered human antibodies or human antibody fragments herein.
6. Multispecific Antibodies
[0117] In certain embodiments, an antibody provided herein is a multispecific antibody, e.g. a bispecific antibody. Multispecific antibodies are monoclonal antibodies that have binding specificities for at least two different sites. In certain embodiments, one of the binding specificities is for HCV E2 protein and the other is for any other antigen. In certain embodiments, bispecific antibodies may bind to two different epitopes of HCV E2 protein. Bispecific antibodies may also be used to localize cytotoxic agents to HCV that expresses HCV E2 protein. Bispecific antibodies can be prepared as full length antibodies or antibody fragments.
[0118] Techniques for making multispecific antibodies include, but are not limited to, recombinant co-expression of two immunoglobulin heavy chain-light chain pairs having different specificities (see Milstein and Cuello, Nature 305: 537 (1983)), WO 93/08829, and Traunecker et al., EMBO J. 10: 3655 (1991)), and "knob-in-hole" engineering (see, e.g., U.S. Patent No. 5,731,168). Multi- specific antibodies may also be made by engineering electrostatic steering effects for making antibody Fc-heterodimeric molecules
(WO 2009/089004A1); cross-linking two or more antibodies or fragments (see, e.g., US Patent No. 4,676,980, and Brennan et al., Science, 229: 81 (1985)); using leucine zippers to produce bi-specific antibodies (see, e.g., Kostelny et al., J. Immunol., 148(5): 1547-1553 (1992)); using "diabody" technology for making bispecific antibody fragments (see, e.g., Hollinger et al., Proc. Natl. Acad. Sci. USA, 90:6444-6448 (1993)); and using single-chain Fv (sFv) dimers (see, e.g. Gruber et al., J. Immunol., 152:5368 (1994)); and preparing trispecific antibodies as described, e.g., in Tutt et al. J. Immunol. 147: 60 (1991).
[0119] Engineered antibodies with three or more functional antigen binding sites, including "Octopus antibodies," are also included herein (see, e.g. US 2006/0025576A1).
[0120] The antibody or fragment herein also includes a "Dual Acting FAb" or "DAF" comprising an antigen binding site that binds to HCV E2 protein as well as another, different antigen (see, US 2008/0069820, for example).
7. Antibody Variants
[0121] In certain embodiments, amino acid sequence variants of the antibodies provided herein are contemplated. For example, it may be desirable to improve the binding affinity
and/or other biological properties of the antibody. Amino acid sequence variants of an antibody may be prepared by introducing appropriate modifications into the nucleotide sequence encoding the antibody, or by peptide synthesis. Such modifications include, for example, deletions from, and/or insertions into and/or substitutions of residues within the amino acid sequences of the antibody. Any combination of deletion, insertion, and substitution can be made to arrive at the final construct, provided that the final construct possesses the desired characteristics, e.g., antigen-binding. a) Substitution, Insertion, and Deletion Variants
[0122] In certain embodiments, antibody variants having one or more amino acid substitutions are provided. Sites of interest for substitutional mutagenesis include the HVRs and FRs. Conservative substitutions are shown in Table A under the heading of
"conservative substitutions." More substantial changes are provided in Table A under the heading of "exemplary substitutions," and as further described below in reference to amino acid side chain classes. Amino acid substitutions may be introduced into an antibody of interest and the products screened for a desired activity, e.g., retained/improved antigen binding, decreased immunogenicity, or improved ADCC or CDC.
Table A

Pro (P) Ala Ala
Ser (S) Thr Thr
Thr (T) Val; Ser Ser
Trp (W) Tyr; Phe Tyr
Tyr (Y) Trp; Phe; Thr; Ser Phe
Val (V) He; Leu; Met; Phe; Ala; Norleucine Leu
[0123] Amino acids may be grouped according to common side-chain properties:
(1) hydrophobic: Norleucine, Met, Ala, Val, Leu, He;
(2) neutral hydrophilic: Cys, Ser, Thr, Asn, Gin;
(3) acidic: Asp, Glu;
(4) basic: His, Lys, Arg;
(5) residues that influence chain orientation: Gly, Pro;
(6) aromatic: Trp, Tyr, Phe.
[0124] Non-conservative substitutions will entail exchanging a member of one of these classes for another class.
[0125] One type of substitutional variant involves substituting one or more hypervariable region residues of a parent antibody (e.g. a humanized or human antibody). Generally, the resulting variant(s) selected for further study will have modifications (e.g., improvements) in certain biological properties (e.g., increased affinity, reduced immunogenicity) relative to the parent antibody and/or will have substantially retained certain biological properties of the parent antibody. An exemplary substitutional variant is an affinity matured antibody, which may be conveniently generated, e.g., using phage display-based affinity maturation techniques such as those described herein. Briefly, one or more HVR residues are mutated and the variant antibodies displayed on phage and screened for a particular biological activity (e.g. binding affinity).
[0126] Alterations (e.g., substitutions) may be made in HVRs, e.g., to improve antibody affinity. Such alterations may be made in HVR "hotspots," i.e., residues encoded by codons that undergo mutation at high frequency during the somatic maturation process (see, e.g., Chowdhury, Methods Mol. Biol. 207: 179-196 (2008)), and/or SDRs (a-CDRs), with the resulting variant VH or VL being tested for binding affinity. Affinity maturation by
constructing and reselecting from secondary libraries has been described, e.g., in Hoogenboom et al. in Methods in Molecular Biology 178: 1-37 (O'Brien et al., ed., Human Press, Totowa, NJ, (2001).) In some embodiments of affinity maturation, diversity is introduced into the variable genes chosen for maturation by any of a variety of methods (e.g., error-prone PCR, chain shuffling, or oligonucleotide-directed mutagenesis). A secondary library is then created. The library is then screened to identify any antibody variants with the desired affinity. Another method to introduce diversity involves HVR-directed approaches, in which several HVR residues (e.g., 4-6 residues at a time) are randomized. HVR residues involved in antigen binding may be specifically identified, e.g., using alanine scanning mutagenesis or modeling. CDR-H3 and CDR-L3 in particular are often targeted.
[0127] In certain embodiments, substitutions, insertions, or deletions may occur within one or more HVRs so long as such alterations do not substantially reduce the ability of the antibody to bind antigen. For example, conservative alterations (e.g., conservative substitutions as provided herein) that do not substantially reduce binding affinity may be made in HVRs. Such alterations may be outside of HVR "hotspots" or SDRs. In certain embodiments of the variant VH and VL sequences provided above, each HVR either is unaltered, or contains no more than one, two or three amino acid substitutions.
[0128] A useful method for identification of residues or regions of an antibody that may be targeted for mutagenesis is called "alanine scanning mutagenesis" as described by
Cunningham and Wells (1989) Science, 244: 1081-1085. In this method, a residue or group of target residues (e.g., charged residues such as arg, asp, his, lys, and glu) are identified and replaced by a neutral or negatively charged amino acid (e.g., alanine or polyalanine) to determine whether the interaction of the antibody with antigen is affected. Further substitutions may be introduced at the amino acid locations demonstrating functional sensitivity to the initial substitutions. Alternatively, or additionally, a crystal structure of an antigen-antibody complex to identify contact points between the antibody and antigen. Such contact residues and neighboring residues may be targeted or eliminated as candidates for substitution. Variants may be screened to determine whether they contain the desired properties.
[0129] Amino acid sequence insertions include amino- and/or carboxyl-terminal fusions ranging in length from one residue to polypeptides containing a hundred or more residues, as well as intrasequence insertions of single or multiple amino acid residues. Examples of terminal insertions include an antibody with an N-terminal methionyl residue. Other insertional variants of the antibody molecule include the fusion to the N- or C-terminus of the
antibody to an enzyme (e.g. for ADEPT) or a polypeptide which increases the serum half-life of the antibody. b) Glycosylation variants
[0130] In certain embodiments, an antibody provided herein is altered to increase or decrease the extent to which the antibody is glycosylated. Addition or deletion of glycosylation sites to an antibody may be conveniently accomplished by altering the amino acid sequence such that one or more glycosylation sites is created or removed.
[0131] Where the antibody comprises an Fc region, the carbohydrate attached thereto may be altered. Native antibodies produced by mammalian cells typically comprise a branched, biantennary oligosaccharide that is generally attached by an N-linkage to Asn297 of the CH2 domain of the Fc region. See, e.g., Wright et al. TIBTECH 15:26-32 (1997). The
oligosaccharide may include various carbohydrates, e.g., mannose, N-acetyl glucosamine (GlcNAc), galactose, and sialic acid, as well as a fucose attached to a GlcNAc in the "stem" of the biantennary oligosaccharide structure. In some embodiments, modifications of the oligosaccharide in an antibody of the invention may be made in order to create antibody variants with certain improved properties.
[0132] In one embodiment, antibody variants are provided having a carbohydrate structure that lacks fucose attached (directly or indirectly) to an Fc region. For example, the amount of fucose in such antibody may be from 1% to 80%, from 1% to 65%, from 5% to 65% or from 20% to 40%. The amount of fucose is determined by calculating the average amount of fucose within the sugar chain at Asn297, relative to the sum of all glycostructures attached to Asn 297 (e. g. complex, hybrid and high mannose structures) as measured by MALDI-TOF mass spectrometry, as described in WO 2008/077546, for example. Asn297 refers to the asparagine residue located at about position 297 in the Fc region (Eu numbering of Fc region residues); however, Asn297 may also be located about + 3 amino acids upstream or downstream of position 297, i.e., between positions 294 and 300, due to minor sequence variations in antibodies. Such fucosylation variants may have improved ADCC function. See, e.g., US Patent Publication Nos. US 2003/0157108 (Presta, L.); US 2004/0093621 (Kyowa Hakko Kogyo Co., Ltd). Examples of publications related to "defucosylated" or "fucose-deficient" antibody variants include: US 2003/0157108; WO 2000/61739; WO 2001/29246; US 2003/0115614; US 2002/0164328; US 2004/0093621; US 2004/0132140; US 2004/0110704; US 2004/0110282; US 2004/0109865; WO 2003/085119; WO
2003/084570; WO 2005/035586; WO 2005/035778; WO2005/053742; WO2002/031140;
Okazaki et al. J. Mol. Biol. 336: 1239-1249 (2004); Yamane-Ohnuki et al. Biotech. Bioeng. 87: 614 (2004). Examples of cell lines capable of producing defucosylated antibodies include Led 3 CHO cells deficient in protein fucosylation (Ripka et al. Arch. Biochem. Biophys. 249:533-545 (1986); US Pat Appl No US 2003/0157108 Al, Presta, L; and WO 2004/056312 Al, Adams et al., especially at Example 11), and knockout cell lines, such as alpha- 1,6- fucosyltransferase gene, FUT8, knockout CHO cells (see, e.g., Yamane-Ohnuki et al.
Biotech. Bioeng. 87: 614 (2004); Kanda, Y. et al., Biotechnol. Bioeng., 94(4):680-688 (2006); and WO2003/085107).
[0133] Antibodies variants are further provided with bisected oligosaccharides, e.g., in which a biantennary oligosaccharide attached to the Fc region of the antibody is bisected by GlcNAc. Such antibody variants may have reduced fucosylation and/or improved ADCC function. Examples of such antibody variants are described, e.g., in WO 2003/011878 (Jean- Mairet et al.); US Patent No. 6,602,684 (Umana et al.); and US 2005/0123546 (Umana et al). Antibody variants with at least one galactose residue in the oligosaccharide attached to the Fc region are also provided. Such antibody variants may have improved CDC function. Such antibody variants are described, e.g., in WO 1997/30087 (Patel et al.); WO 1998/58964 (Raju, S.); and WO 1999/22764 (Raju, S.).
[0134] In some embodiments, an anti-HCV antibody glycosylation variant comprises a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23, and a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25. c) Fc region variants
[0135] In certain embodiments, one or more amino acid modifications may be introduced into the Fc region of an antibody provided herein, thereby generating an Fc region variant. The Fc region variant may comprise a human Fc region sequence (e.g., a human IgGl, IgG2, IgG3 or IgG4 Fc region) comprising an amino acid modification (e.g. a substitution) at one or more amino acid positions.
[0136] In certain embodiments, the invention contemplates an antibody variant that possesses some but not all effector functions, which make it a desirable candidate for applications in which the half life of the antibody in vivo is important yet certain effector functions (such as complement and ADCC) are unnecessary or deleterious. In vitro and/or in vivo cytotoxicity assays can be conducted to confirm the reduction/depletion of CDC and/or ADCC activities. For example, Fc receptor (FcR) binding assays can be conducted to ensure that the antibody lacks FcyR binding (hence likely lacking ADCC activity), but retains FcRn
binding ability. The primary cells for mediating ADCC, NK cells, express Fc(RIII only, whereas monocytes express Fc(RI, Fc(RII and Fc(RIII. FcR expression on hematopoietic cells is summarized in Table 3 on page 464 of Ravetch and Kinet, Annu. Rev. Immunol.
9:457-492 (1991). Non-limiting examples of in vitro assays to assess ADCC activity of a molecule of interest is described in U.S. Patent No. 5,500,362 (see, e.g. Hellstrom, I. et al. Proc. Nat'lAcad. Sci. USA 83:7059-7063 (1986)) and Hellstrom, I et al., Proc. Nat'lAcad. Sci. USA 82: 1499-1502 (1985); 5,821,337 (see Bruggemann, M. et al., J. Exp. Med.
166: 1351-1361 (1987)). Alternatively, non-radioactive assays methods may be employed (see, for example, ACTI™ non-radioactive cytotoxicity assay for flow cytometry
(CellTechnology, Inc. Mountain View, CA; and CytoTox 96® non-radioactive cytotoxicity assay (Promega, Madison, WI). Useful effector cells for such assays include peripheral blood mononuclear cells (PBMC) and Natural Killer (NK) cells. Alternatively, or additionally, ADCC activity of the molecule of interest may be assessed in vivo, e.g., in an animal model such as that disclosed in Clynes et al. Proc. Nat'lAcad. Sci. USA 95:652-656 (1998). Clq binding assays may also be carried out to confirm that the antibody is unable to bind Clq and hence lacks CDC activity. See, e.g., Clq and C3c binding ELISA in WO 2006/029879 and WO 2005/100402. To assess complement activation, a CDC assay may be performed (see, for example, Gazzano-Santoro et al., J. Immunol. Methods 202: 163 (1996); Cragg, M.S. et al., Blood 101: 1045-1052 (2003); and Cragg, M.S. and M.J. Glennie, Blood 103:2738-2743 (2004)). FcRn binding and in vivo clearance/half life determinations can also be performed using methods known in the art (see, e.g., Petkova, S.B. et al., Int'l. Immunol. 18(12): 1759- 1769 (2006)).
[0137] Antibodies with reduced effector function include those with substitution of one or more of Fc region residues 238, 265, 269, 270, 297, 327 and 329 (U.S. Patent No. 6,737,056). Such Fc mutants include Fc mutants with substitutions at two or more of amino acid positions 265, 269, 270, 297 and 327, including the so-called "DANA" Fc mutant with substitution of residues 265 and 297 to alanine (US Patent No. 7,332,581).
[0138] Certain antibody variants with improved or diminished binding to FcRs are described. (See, e.g., U.S. Patent No. 6,737,056; WO 2004/056312, and Shields et al., /. Biol. Chem. 9(2): 6591-6604 (2001).)
[0139] In certain embodiments, an antibody variant comprises an Fc region with one or more amino acid substitutions which improve ADCC, e.g., substitutions at positions 298, 333, and/or 334 of the Fc region (EU numbering of residues).
[0140] In some embodiments, alterations are made in the Fc region that result in altered (i.e., either improved or diminished) Clq binding and/or Complement Dependent
Cytotoxicity (CDC), e.g., as described in US Patent No. 6,194,551, WO 99/51642, and Idusogie et al. J. Immunol. 164: 4178-4184 (2000).
[0141] Antibodies with increased half lives and improved binding to the neonatal Fc receptor (FcRn), which is responsible for the transfer of maternal IgGs to the fetus (Guyer et al., J. Immunol. 117:587 (1976) and Kim et al., J. Immunol. 24:249 (1994)), are described in US2005/0014934A1 (Hinton et al.). Those antibodies comprise an Fc region with one or more substitutions therein which improve binding of the Fc region to FcRn. Such Fc variants include those with substitutions at one or more of Fc region residues: 238, 256, 265, 272, 286, 303, 305, 307, 311, 312, 317, 340, 356, 360, 362, 376, 378, 380, 382, 413, 424 or 434, e.g., substitution of Fc region residue 434 (US Patent No. 7,371,826).
[0142] See also Duncan & Winter, Nature 322:738-40 (1988); U.S. Patent No. 5,648,260; U.S. Patent No. 5,624,821; and WO 94/29351 concerning other examples of Fc region variants. d) Cysteine engineered antibody variants
[0143] In certain embodiments, it may be desirable to create cysteine engineered antibodies, e.g., "thioMAbs," in which one or more residues of an antibody are substituted with cysteine residues. In particular embodiments, the substituted residues occur at accessible sites of the antibody. By substituting those residues with cysteine, reactive thiol groups are thereby positioned at accessible sites of the antibody and may be used to conjugate the antibody to other moieties, such as drug moieties or linker-drug moieties, to create an immunoconjugate, as described further herein. In certain embodiments, any one or more of the following residues may be substituted with cysteine: V205 (Kabat numbering) of the light chain; Al 18 (EU numbering) of the heavy chain; and S400 (EU numbering) of the heavy chain Fc region. Cysteine engineered antibodies may be generated as described, e.g., in U.S. Patent No. 7,521,541. e) Antibody Derivatives
[0144] In certain embodiments, an antibody provided herein may be further modified to contain additional nonproteinaceous moieties that are known in the art and readily available. The moieties suitable for derivatization of the antibody include but are not limited to water soluble polymers. Non-limiting examples of water soluble polymers include, but are not
limited to, polyethylene glycol (PEG), copolymers of ethylene glycol/propylene glycol, carboxymethylcellulose, dextran, polyvinyl alcohol, polyvinyl pyrrolidone, poly-1, 3- dioxolane, poly-1, 3, 6-trioxane, ethylene/maleic anhydride copolymer, polyaminoacids (either homopolymers or random copolymers), and dextran or poly(n-vinyl pyrrolidone)polyethylene glycol, propropylene glycol homopolymers, prolypropylene oxide/ethylene oxide copolymers, polyoxyethylated polyols (e.g., glycerol), polyvinyl alcohol, and mixtures thereof. Polyethylene glycol propionaldehyde may have advantages in manufacturing due to its stability in water. The polymer may be of any molecular weight, and may be branched or unbranched. The number of polymers attached to the antibody may vary, and if more than one polymer are attached, they can be the same or different molecules. In general, the number and/or type of polymers used for derivatization can be determined based on considerations including, but not limited to, the particular properties or functions of the antibody to be improved, whether the antibody derivative will be used in a therapy under defined conditions, etc.
[0145] In another embodiment, conjugates of an antibody and nonproteinaceous moiety that may be selectively heated by exposure to radiation are provided. In one embodiment, the nonproteinaceous moiety is a carbon nanotube (Kam et al., Proc. Natl. Acad. Sci. USA 102: 11600-11605 (2005)). The radiation may be of any wavelength, and includes, but is not limited to, wavelengths that do not harm ordinary cells, but which heat the nonproteinaceous moiety to a temperature at which cells proximal to the antibody-nonproteinaceous moiety are killed.
B. Recombinant Methods and Compositions
[0146] The invention also provides methods of producing anti-HCV antibodies using recombinant techniques. For example, polypeptides can be prepared using isolated nucleic acids encoding such antibodies or fragments thereof, vectors and host-cells comprising such nucleic acids.
[0147] Antibodies may be produced using recombinant methods and compositions, e.g., as described in U.S. Patent No. 4,816,567. In one embodiment, isolated nucleic acid encoding an anti-HCV antibody described herein is provided. Such nucleic acid may encode an amino acid sequence comprising the VL and/or an amino acid sequence comprising the VH of the antibody (e.g., the light and/or heavy chains of the antibody). In some embodiments, the isolated nucleic acid encodes a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23. In some embodiments, the isolated nucleic acid encodes a VL amino
acid sequence selected from the group consisting of SEQ ID NOs:24 and 25. In some embodiments, the isolated nucleic acid encodes a heavy chain amino acid sequence selected from the group consisting of SEQ ID NOs:27 and 28. In some embodiments, the isolated nucleic acid encodes a light chain amino acid sequence of SEQ ID NO:30.
[0148] For recombinant production of anti-HCV antibodies or fragments thereof, nucleic acids encoding the desired antibodies or antibody fragments as described herein, are isolated and inserted into a replicable vector for further cloning (amplification of the DNA) or for expression. In a further embodiment, one or more vectors (e.g., expression vectors) comprising such nucleic acid are provided. In some embodiments, a vector comprises a nucleic acid encoding a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23. In some embodiments, a vector comprises a nucleic acid encoding a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25. In other embodiments, a vector comprises a nucleic acid encoding a heavy chain amino acid sequence selected from the group consisting of SEQ ID NOs:27 and 28. In still other embodiments, a vector comprises a nucleic acid encoding a light chain amino acid sequence of SEQ ID NO:30. DNA encoding the polyclonal or monoclonal antibodies is readily isolated (e.g., with oligonucleotide probes that specifically bind to genes encoding the heavy and light chains of the antibody) and sequenced using conventional procedures. Many cloning and/or expression vectors are commercially available. Vector components generally include, but are not limited to, one or more of the following, a signal sequence, an origin of replication, one or more marker genes, a multiple cloning site containing recognition sequences for numerous restriction endonucleases, an enhancer element, a promoter, and a transcription termination sequence.
Signal sequence component
[0149] The antibodies or fragments thereof may be produced recombinantly not only directly, but also as a fusion protein, where the antibody is fused to a heterologous polypeptide, preferably a signal sequence or other polypeptide having a specific cleavage site at the N-terminus of the mature protein or polypeptide. The heterologous signal sequence selected preferably is one that is recognized and processed (i.e., cleaved by a signal peptidase) by eukaryotic host-cells. For prokaryotic host-cells that do not recognize and process native mammalian signal sequences, the eukaryotic (i.e., mammalian) signal sequence is replaced by a prokaryotic signal sequence selected, for example, from the group consisting of leader sequences from alkaline phosphatase, penicillinase, lpp, or heat-stable enterotoxin II genes. For yeast secretion the native signal sequence may be substituted by,
e.g., the yeast invertase leader, factor leader (including Saccharomyces and Kluyveromyces - factor leaders), or acid phosphatase leader, the C. albicans glucoamylase leader, or the signal described in WO 90/13646. In mammalian cell expression, mammalian signal sequences as well as viral secretory leaders, for example, the herpes simplex virus gD signal, are available.
[0150] The DNA for such precursor region is ligated in reading frame to the DNA encoding the antibodies or fragments thereof.
Origin of replication
[0151] Both expression and cloning vectors contain a nucleic acid sequence that enables the vector to replicate in one or more selected host-cells. Generally, in cloning vectors this sequence is one that enables the vector to replicate independently of the host chromosomal DNA, and includes origins of replication or autonomously replicating sequences. Such sequences are well known for a variety of bacteria, yeast, and viruses. The origin of replication from the plasmid pBR322 is suitable for most Gram-negative bacteria, the 2μ plasmid origin is suitable for yeast, and various viral origins (SV40, polyoma, adenovirus, vesicular stomatitis virus ("VSV") or bovine papilloma virus ("BPV") are useful for cloning vectors in mammalian cells. Generally, the origin of replication component is not needed for mammalian expression vectors (the SV40 origin may typically be used only because it contains the early promoter).
Selection gene component
[0152] Expression and cloning vectors may also contain a selection gene, known as a selectable marker. Typical selection genes encode proteins that (a) confer resistance to antibiotics or other toxins, e.g., ampicillin, neomycin, methotrexate, or tetracycline, (b) complement auxotrophic deficiencies, or (c) supply critical nutrients not available from complex media, e.g., the gene encoding D-alanine racemase for Bacilli.
[0153] One example of a selection scheme utilizes a drug to arrest growth of a host-cell. Those cells that are successfully transformed with a heterologous gene produce a protein conferring drug resistance and thus survive the selection regimen. Examples of such dominant selection strategies use the drugs neomycin, mycophenolic acid and hygromycin.
[0154] Another example of suitable selectable markers for mammalian cells are those that enable the identification of cells competent to take up the antibody- or antibody fragment- encoding nucleic acids, such as dihydrofolate reductase ("DHFR"), thymidine kinase, metallothionein-I and -II, preferably primate metallothionein genes, adenosine deaminase, ornithine decarboxylase, and the like.
[0155] For example, cells transformed with the DHFR selection gene are first identified by culturing all of the transformants in a culture medium that contains methotrexate (Mtx), a competitive antagonist of DHFR. An exemplary host-cell strain for use with wild-type DHFR is the Chinese hamster ovary ("CHO") cell line lacking DHFR activity (e.g., ATCC CRL-9096).
[0156] Another example of suitable selectable markers for mammalian cells are those that enable the identification of cells competent to take up the antibody- or antibody fragment- encoding nucleic acids, such as dihydrofolate reductase ("DHFR"), glutamine synthetase (GS), thymidine kinase, metallothionein-I and -II, preferably primate metallothionein genes, adenosine deaminase, ornithine decarboxylase, and the like.
[0157] Alternatively, cells transformed with the GS (glutamine synthetase) gene are identified by culturing the transformants in a culture medium containing L-methionine sulfoximine (Msx), an inhibitor of GS. Under these conditions, the GS gene is amplified along with any other co-transformed nucleic acid. The GS selection/amplification system may be used in combination with the DHFR selection/amplification system described above.
[0158] Alternatively, host-cells (particularly wild-type hosts that contain endogenous DHFR) transformed or co-transformed with DNA sequences encoding anti-CD83 agonist antibodies or fragments thereof, wild-type DHFR protein, and another selectable marker such as aminoglycoside 3'-phosphotransferase ("APH") can be selected by cell growth in medium containing a selection agent for the appropriate selectable marker, such as an aminoglycosidic antibiotic, e.g., kanamycin, neomycin, or G418. See U.S. Patent No. 4,965,199.
[0159] A suitable selection gene for use in yeast is the trpl gene present in the yeast plasmid YRp7 (Stinchcomb et al., Nature, 282:39 (1979)). The trpl gene provides a selection marker for a mutant strain of yeast lacking the ability to grow medium containing tryptophan (e.g., ATCC No. 44076 or PEP4-1). Jones, Genetics, 85: 12 (1977). The presence of the trpl lesion in the yeast host-cell genome then provides an effective environment for detecting transformation by growth in the absence of tryptophan. Similarly, Lew2-deficient yeast strains (e.g., ATCC 20,622 or 38,626) can be complemented by known plasmids bearing the Leu2 gene.
[0160] In addition, vectors derived from the 1.6 μιη circular plasmid pKDl can be used for transformation of Kluyveromyces yeasts. Alternatively, an expression system for large-scale production of recombinant calf chymosin was reported for K. lactis. Van den Berg,
Bio/Technology, 8: 135 (1990). Stable multi-copy expression vectors for secretion of mature
recombinant human serum albumin by industrial strains of Kluyveromyces have also been disclosed. Fleer et al., Bio/Technology, 9:968-975 (1991).
Promoter component
[0161] Expression and cloning vectors usually contain a promoter that is recognized by the host organism and is operably linked to the nucleic acid encoding the anti-HCV antibodies or fragments thereof. Promoters suitable for use with prokaryotic hosts include the phoA promoter, lactamase and lactose promoter systems, alkaline phosphatase promoter, a tryptophan promoter system, and hybrid promoters such as the tac promoter, although other known bacterial promoters are also suitable. Promoters for use in bacterial systems also will contain a Shine-Dalgarno (S.D.) sequence operably linked to the DNA encoding the antibodies and antibody fragments.
[0162] Promoter sequences are known for eukaryotes. Virtually all eukaryotic genes have an AT-rich region located approximately 25 to 30 bases upstream from the site where transcription is initiated. Another sequence found 70 to 80 bases upstream from the start of transcription of many genes is a CNCAAT region where N may be any nucleotide. At the 3' end of most eukaryotic genes is an AATAAA sequence that may be the signal for addition of the polyA tail to the 3' end of the coding sequence. All of these sequences may be inserted into eukaryotic expression vectors.
[0163] Examples of suitable promoter sequences for use with yeast hosts include the promoters for 3-phosphoglycerate kinase or other glycolytic enzymes, such as enolase, glyceraldehyde-3-phosphate dehydrogenase, hexokinase, pyruvate decarboxylase, phospho-fructokinase, glucose-6-phosphate isomerase, 3-phosphoglycerate mutase, pyruvate kinase, triosephosphate isomerase, phosphoglucose isomerase, and glucokinase.
[0164] Inducible promoters in yeast have the additional advantage of permitting
transcription controlled by growth conditions. Exemplary inducible promoters include the promoter regions for alcohol dehydrogenase 2, isocytochrome C, acid phosphatase, degradative enzymes associated with nitrogen metabolism, metallothionein, glyceraldehyde- 3-phosphate dehydrogenase, and enzymes responsible for maltose and galactose utilization. Suitable vectors and promoters for use in yeast expression are further described in EP 73,657. Yeast enhancers also are advantageously used with yeast promoters.
[0165] Transcription of nucleic acids encoding antibodies or fragments thereof from vectors in mammalian host-cells can be controlled, for example, by promoters obtained from the genomes of viruses such as polyoma virus, fowlpox virus, adenovirus (such as
Adenovirus 2), bovine papilloma virus, avian sarcoma virus, cytomegalovirus, a retrovirus,
hepatitis-B virus and most preferably Simian Virus 40 (SV40), by heterologous mammalian promoters, e.g., the actin promoter or an immunoglobulin promoter, and by heat-shock gene promoters, provided such promoters are compatible with the desired host-cell systems.
[0166] The early and late promoters of the SV40 virus are conveniently obtained as an SV40 restriction fragment that also contains the SV40 viral origin of replication. The immediate early promoter of the human cytomegalovirus is conveniently obtained as a Hindlll E restriction fragment. A system for expressing DNA in mammalian hosts using the bovine papilloma virus as a vector is disclosed in U.S. Patent No. 4,419,446. A modification of this system is described in U.S. Patent No. 4,601,978. See also Reyes et al., Nature 297:598-601 (1982), regarding methods for expression of human interferon cDNA in mouse cells under the control of a thymidine kinase promoter from herpes simplex virus.
Alternatively, the Rous Sarcoma Virus long terminal repeat can be used as the promoter.
Enhancer element component
[0167] Transcription of a DNA encoding the antibodies or fragments thereof by higher eukaryotes is often increased by inserting an enhancer sequence into the vector. Many enhancer sequences are now known from mammalian genes (globin, elastase, albumin, a- fetoprotein, and insulin). Typically, however, one of ordinary skill in the art will use an enhancer from a eukaryotic virus. Examples include the SV40 enhancer on the late side of the replication origin (bp 100-270), the cytomegalovirus early promoter enhancer, the polyoma enhancer on the late side of the replication origin, and adenovirus enhancers. See also Yaniv, Nature 297: 17-18 (1982) on enhancing elements for activation of eukaryotic promoters. The enhancer may be spliced into the vector at a position 5' or 3' to the antibody- or antibody-fragment encoding sequences, but is preferably located at a site 5' of the promoter.
Transcription termination component
[0168] Expression vectors used in eukaryotic host-cells (yeast, fungi, insect, plant, animal, human, or nucleated cells from other multicellular organisms) will also contain sequences necessary for the termination of transcription and for stabilizing the mRNA. Such sequences are commonly available from the 5' and, occasionally 3', untranslated regions of eukaryotic or viral DNAs or cDNAs. These regions contain nucleotide segments transcribed as
polyadenylated fragments in the untranslated portion of the mRNA encoding antibodies or fragments thereof. One useful transcription termination component is the bovine growth hormone polyadenylation region. See W094/11026 and the expression vector disclosed therein.
Selection and transformation of host-cells
[0169] Suitable host cells for cloning or expressing nucleic acid encoding anti-HCV antibodies or fragments thereof in the vectors described include prokaryotic or eukaryotic cells described herein. For example, antibodies may be produced in bacteria, in particular when glycosylation and Fc effector function are not needed. For expression of antibody fragments and polypeptides in bacteria, see, e.g., U.S. Patent Nos. 5,648,237, 5,789,199, and 5,840,523. (See also Charlton, Methods in Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ, 2003), pp. 245-254, describing expression of antibody fragments in E. coli.) After expression, the antibody may be isolated from the bacterial cell paste in a soluble fraction and can be further purified.
[0170] In addition to prokaryotes, eukaryotic microbes such as filamentous fungi or yeast are suitable cloning or expression hosts for antibody-encoding vectors, including fungi and yeast strains whose glycosylation pathways have been "humanized," resulting in the production of an antibody with a partially or fully human glycosylation pattern. See
Gerngross, Nat. Biotech. 22: 1409-1414 (2004), and Li et al., Nat. Biotech. 24:210-215 (2006).
[0171] Suitable host cells for the expression of glycosylated antibody are also derived from multicellular organisms (invertebrates and vertebrates). Examples of invertebrate cells include plant and insect cells. Numerous baculoviral strains have been identified which may be used in conjunction with insect cells, particularly for transfection of Spodoptera frugiperda cells.
[0172] Plant cell cultures can also be utilized as hosts. See, e.g., US Patent Nos. 5,959,177, 6,040,498, 6,420,548, 7,125,978, and 6,417,429 (describing PLANTIBODIES™ technology for producing antibodies in transgenic plants).
[0173] Vertebrate cells may also be used as hosts. For example, mammalian cell lines that are adapted to grow in suspension may be useful. Other examples of useful mammalian host cell lines are monkey kidney CV1 line transformed by SV40 (COS-7); human embryonic kidney line (293 or 293 cells as described, e.g., in Graham et al., /. Gen Virol. 36:59 (1977)); baby hamster kidney cells (BHK); mouse Sertoli cells (TM4 cells as described, e.g., in Mather, Biol. Reprod. 23:243-251 (1980)); monkey kidney cells (CV1); African green monkey kidney cells (VERO-76); human cervical carcinoma cells (HELA); canine kidney cells (MDCK; buffalo rat liver cells (BRL 3A); human lung cells (W138); human liver cells (Hep G2); mouse mammary tumor (MMT 060562); TRI cells, as described, e.g., in Mather et al., Annals N.Y. Acad. Sci. 383:44-68 (1982); MRC 5 cells; and FS4 cells. Other useful
mammalian host cell lines include Chinese hamster ovary (CHO) cells, including DHFR" CHO cells (Urlaub et al, Pwc. Natl. Acad. Sci. USA 77:4216 (1980)); and myeloma cell lines such as Y0, NSO and Sp2/0. For a review of certain mammalian host cell lines suitable for antibody production, see, e.g., Yazaki and Wu, Methods in Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ), pp. 255-268 (2003).
Culturing the host-cells
[0174] In some embodiments, host-cells are transformed with the above-described expression or cloning vectors for anti-HCV antibody or antibody fragment production are cultured in conventional nutrient media modified as appropriate for inducing promoters, selecting transformants, or amplifying the genes encoding the desired sequences.
Commercially available media such as Ham's F10 (Sigma), Minimal Essential Medium ((MEM), Sigma), RPMI-1640 (Sigma), and Dulbecco's Modified Eagle's Medium ((DMEM), Sigma) are suitable for culturing the host-cells. In addition, any of the media described in Ham et al., Meth. Enz. 58:44 (1979), Barnes et al., Anal. Biochem. l02:255 (1980), U.S.
Patent Nos. 4,767,704; 4,657,866; 4,927,762; 4,560,655; or 5,122,469; WIPO Publication Nos. WO 90/03430; WO 87/00195; or U.S. Patent Re. 30,985 may be used as culture media for the host-cells. Any of these media may be supplemented as necessary with hormones and/or other growth factors (such as insulin, transferrin, or epidermal growth factor), salts (such as sodium chloride, calcium, magnesium, and phosphate), buffers (such as HEPES), nucleotides (such as adenosine and thymidine), antibiotics (such as GENTAMYCIN™ drug), trace elements (defined as inorganic compounds usually present at final concentrations in the micromolar range), and glucose or an equivalent energy source. Any other necessary supplements may also be included at appropriate concentrations that would be known to those skilled in the art. The culture conditions, such as temperature, pH, and the like, are those previously used with the host-cell selected for expression, and will be apparent to the ordinarily skilled artisan.
[0175] In some embodiments, a host cell comprising one or more nucleic acid encoding an anti-HCV antibody or fragment thereof as described herein is provided. In one such embodiment, a host cell comprises (e.g., has been transformed with): (1) a vector comprising a nucleic acid that encodes an amino acid sequence comprising the VL of the antibody and an amino acid sequence comprising the VH of the antibody, or (2) a first vector comprising a nucleic acid that encodes an amino acid sequence comprising the VL of the antibody and a second vector comprising a nucleic acid that encodes an amino acid sequence comprising the VH of the antibody. In one embodiment, the host cell is eukaryotic, e.g. a Chinese Hamster
Ovary (CHO) cell or lymphoid cell (e.g., Y0, NSO, Sp20 cell). In some embodiments, a host cell comprises a nucleic acid encoding a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23. In other embodiments, a host cell comprises a nucleic acid encoding a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25. In other embodiments, a host cell comprises a nucleic acid encoding a heavy chain amino acid sequence selected from the group consisting of SEQ ID NOs:27 and 28. In still other embodiments, a host cell comprises a nucleic acid encoding a light chain amino acid sequence of SEQ ID NO:30. In one embodiment, a method of making an anti-HCV antibody is provided, wherein the method comprises culturing a host cell comprising a nucleic acid encoding the antibody, as provided above, under conditions suitable for expression of the antibody, and optionally recovering the antibody from the host cell (or host cell culture medium). In some embodiments, the host cell is a 293T cell.
[0176] In some embodiments, an antibody that binds HCV E2 protein is produced by a method comprising culturing a host cell comprising one or more nucleic acid encoding an antibody described herein, under a condition suitable for expression of the one or more nucleic acid, and recovering the antibody produced by the cell. In a further embodiment, the one or more nucleic acid encodes a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23. In another further embodiment, the one or more nucleic acid encodes a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25. In a further embodiment, the one or more nucleic acid encodes a heavy chain amino acid sequence selected from the group consisting of SEQ ID NOs:27 and 28. In still another further embodiment, the one or more nucleic acid encodes a light chain amino acid sequence of SEQ ID NO:30. In some embodiments, the antibody that binds HCV E2 protein produced by a method comprising culturing a host cell comprising one or more nucleic acid encoding an antibody described herein has a lysine residue removed from the C- terminus. In some embodiments, the host cell is a 293T cell.
Purification of antibody
[0177] When using recombinant techniques, the anti-HCV antibodies or antibody fragments can be produced intracellularly, in the periplasmic space, or secreted directly into the medium. If the antibodies are produced intracellularly, as a first step, the particulate debris from either host-cells or lysed fragments is removed, for example, by centrifugation or ultrafiltration. Carter et al., Bio/Technology 10: 163-167 (1992) describe a procedure for isolating antibodies which are secreted to the periplasmic space of E. coli. Briefly, cell paste is thawed in the presence of sodium acetate (pH 3.5), EDTA, and
phenylmethylsulfonylfhioride (PMSF) over about 30 minutes. Cell debris can be removed by centrifugation. Where the antibody is secreted into the medium, supernatants from such expression systems are generally first concentrated using a commercially available protein concentration filter, for example, an Amicon or Millipore Pellicon ultrafiltration unit. A protease inhibitor such as PMSF may be included in any of the foregoing steps to inhibit proteolysis and antibiotics may be included to prevent the growth of adventitious
contaminants.
[0178] The antibody or antibody fragment compositions prepared from such cells can be purified using, for example, hydroxylapatite chromatography, gel electrophoresis, dialysis, and affinity chromatography, with affinity chromatography being the preferred purification technique. The suitability of protein A as an affinity ligand depends on the species and isotype of any immunoglobulin Fc domain that is present in the antibody. Protein A can be used to purify antibodies or antibody fragments that are based on human 1, 2, or 4 heavy chains (Lindmark et al., J. Immunol. Meth. 62: 1-13 (1983)). Protein G is recommended for all mouse isotypes and for human 3 heavy chain antibodies or antibody fragments (Guss et al., EMBO J. 5: 15671575 (1986)). The matrix to which the affinity ligand is attached is most often agarose, but other matrices are available. Mechanically stable matrices such as controlled pore glass or poly(styrene-divinyl)benzene allow for faster flow rates and shorter processing times than can be achieved with agarose. Where the antibodies or antibody fragments comprise a CH3 domain, the Bakerbond ABX™resin (J. T. Baker, Phillipsburg, NJ) is useful for purification. Other techniques for protein purification, such as fractionation on an ion-exchange column, ethanol precipitation, Reverse Phase HPLC, chromatography on silica, heparin, SEPHAROSE™, or anion or cation exchange resins (such as a polyaspartic acid column), as well as chromatofocusing, SDS-PAGE, and ammonium sulfate precipitation are also available depending on the antibody or antibody fragment to be recovered.
[0179] Following any preliminary purification step or steps, the mixture comprising the antibody or antibody fragment of interest and contaminants may be subjected to low pH hydrophobic interaction chromatography using an elution buffer at a pH between about 2.5- 4.5, preferably performed at low salt concentrations {e.g., from about 0-0.25 M salt).
[0180] In general, various methodologies for preparing antibodies for use in research, testing, and clinical applications are well-established in the art, consistent with the above- described methodologies and/or as deemed appropriate by one skilled in the art for a particular antibody of interest.
C. Assays
[0181] Anti-HCV antibodies provided herein may be identified, screened for, or characterized for their physical/chemical properties and/or biological activities by various assays known in the art. Anti-HCV antibodies with certain biological characteristics may be selected as described in the Examples.
1. Binding assays and other assays
[0182] In one aspect, an anti-HCV antibody of the invention is tested for its antigen binding activity, e.g., by known methods such as ELISA, Western blot, etc.
[0183] In another aspect, competition assays may be used to identify an anti-HCV antibody that competes with any anti-HCV antibody described herein (e.g., V362) for binding to HCV E2 protein. In certain embodiments, competitive binding may be determined using an Enzyme-Linked Immunosorbent Assay (ELISA) assay. For example, in certain
embodiments, an antibody is provided that binds to HCV E2 protein competitively with an anti-HCV antibody comprising a VH amino acid sequence selected from the group consisting of SEQ ID NOs: 17-23, and a VL amino acid sequence selected from the group consisting of SEQ ID NOs:24 and 25. In certain embodiments, an antibody is provided that binds to HCV E2 protein competitively with an anti-HCV antibody comprising the heavy chain amino acid sequence shown in SEQ ID NOs:27 or 28 and the light chain amino acid sequence shown in SEQ ID NO:30. In certain embodiments, such a competing antibody binds to the same epitope (e.g., a linear or a conformational epitope) that is bound by any anti-HCV antibody described herein (e.g., V362). Detailed exemplary methods for mapping an epitope to which an antibody binds are provided in Morris (1996) "Epitope Mapping Protocols," in Methods in Molecular Biology vol. 66 (Humana Press, Totowa, NJ).
[0184] In an exemplary competition assay, immobilized HCV E2 protein is incubated in a solution comprising a first labeled antibody that binds to HCV E2 protein (e.g., V362) and a second unlabeled antibody that is being tested for its ability to compete with the first antibody for binding to HCV E2 protein. The second antibody may be present in a hybridoma supernatant. As a control, immobilized HCV E2 protein is incubated in a solution comprising the first labeled antibody but not the second unlabeled antibody. After incubation under conditions permissive for binding of the first antibody to HCV E2 protein, excess unbound antibody is removed, and the amount of label associated with immobilized HCV E2 protein is measured. If the amount of label associated with immobilized HCV E2 protein is
substantially reduced in the test sample relative to the control sample, then that indicates that
the second antibody is competing with the first antibody for binding to HCV E2 protein. See Harlow and Lane (1988) Antibodies: A Laboratory Manual ch.14 (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY).
2. Activity assays
[0185] In one aspect, assays are provided for identifying anti-HCV antibodies as described herein having biological activity. Biological activity may include, e.g., neutralization of HCV pseudoparticles (HCVpp), neutralization of infectious cell culture HCV (HCVcc), inhibition of HCV infection, etc. Antibodies having such biological activity in vivo and/or in vitro are also provided.
[0186] In certain embodiments, an antibody of the invention is tested for such biological activity. Biological activity can be expressed as 90% effective concentration (EC90) values as described in Hotzel et al., Protein Eng Des Sel. (2011), 24:679-89 and Diao et al., J Virol. (2012), 86(20): 10935-49. In some embodiments, antibodies can also be screen for their ability to neutralize an HCV infection. In some embodiments, neutralization of an HCV infection is based on a HCV pseudotyped particles (HCVpp) neutralization assay as described herein. For example, neutralization of genotype 2a E2 protein-expressing HCVpp by anti- HCV antibodies is analyzed using a neutralization assay with HCVpp stocks expressing luciferase. About 5x10 Huh-7.5 cells are seeded in a 96- well plate 16 hours prior to the neutralization assay. The following day, three-fold dilutions of anti-HCV antibody are incubated with naive Huh-7.5 cells for 1 hour at 37°C, followed by the addition of HCVpp for an additional 4 hours. Unbound virus is removed by replacing with fresh DMEM and the cells are maintained in culture for 3 days before measuring HCV infection. HCVpp infection is determined using the Luciferase Assay System (Promega) which detects the luciferase expressed by HCVpp and allows measurement of inhibition of HCV infection and spread for calculation of the anti-HCV antibody EC90 value.
[0187] In some embodiments, neutralization of an HCV infection is based on a
recombinant cell culture-derived HCV (HCVcc) neutralization assay infecting human hepatoma cell lines as described herein. For example, neutralization of HCVcc expressing wild-type genotype 2a Jcl is conducted as previously described in Hotzel et al., Protein Eng Des Sel. (2011), 24:679-89 and Diao et al., J Virol. (2012), 86(20): 10935-49, to test the ability of the anti-HCV antibodies to inhibit entry and infection of HCV. Multiplicity of infection (MOI) for Jcl HCVcc is calculated based on virus titer as measured by TCID50 calculations for obtainment of an EC90 value.
[0188] In some embodiments, an anti-HCV antibody provided herein has an EC90 of < 500 μg/mL, < 250 μg/mL, < 100 μg/mL, < 10 μg/mL, < 1 μg/mL, < 0.1 μg/mL, < 0.01 μg/mL, or < 0.001 μg/mL.. In some embodiments herein, an anti-HCV antibody provided herein has an EC90 of less than about any of about 100 μg/mL, 95 μg/mL, 90 μg/mL, 85 μg/mL, 80 μg/mL, 75 μg/mL, 70 μg/mL, 65 μg/mL, 60 μg/mL, 55 μg/mL, 50 μg/mL, 45 μg/mL, 40 μg/mL, 35 μg/mL, 30 μg/mL, 25 μg/mL, 20 μg/mL, 15 μg/mL, 10 μg/mL, 9 μg/mL, 8 μg/mL, 7 μg/mL, 6 μg/mL, 5 μg/mL, 4 μg/mL, 3 μg/mL, 2 μg/mL, 1 μg/mL, 0.9 μg/mL, 0.8 μg/mL, 0.7 μg/mL, 0.6 μg/mL, 0.5 μg/mL, 0.4 μg/mL, 0.3 μg/mL, 0.2 μg/mL, 0.1 μg/mL, 0.09 μg/mL, 0.08 μg/mL, 0.07 μg/mL, 0.06 μg/mL, 0.05 μg/mL, 0.04 μg/mL, 0.03 μg/mL, 0.02 μg/mL, 0.01 μg/mL, 0.009 μg/mL, 0.008 μg/mL, 0.007 μg/mL, 0.006 μg/mL, 0.005 μg/mL, 0.004 μg/mL, 0.003 μg/mL, 0.002 μg/mL, or 0.001 μg/mL, inclusive, including any values in between these numbers, for inhibition of infection by an HCV genotype selected from the group consisting of genotype 1 (e.g., genotype la and genotype lb), genotype 2 (e.g. , genotype 2a, genotype 2b, genotype 2c), genotype 3 (e.g., genotype 3a), genotype 4 (e.g., genotype 4a), genotype 5, and genotype 6. In some embodiments, an anti-HCV antibody described herein has a lower EC90 (for example as measured in an HCVpp or HCVcc assay described herein) as compared to the EC90 of a parent antibody (such as MRCTlO. l) described herein for inhibition of HCV infection. In some embodiments, the antibody has an EC90 of less than about 5 μg/ml, less than about 4 μg/ml, less than about 3 μg/ml, less than about 2 μg/ml, or about 1 μg/ml for inhibition of infection of an HCV genotype. In some embodiments, the EC90 value of an anti-HCV antibody as described herein is less than any of 50%, 40%, 30%, 20%, 15%, 10%, 5%, or 1%, inclusive, including any value in between these numbers, of the EC90 value of MRCTlO. l for inhibition of infection by an HCV genotype selected from the group consisting of genotype 1 (e.g., genotype la and genotype lb), genotype 2 (e.g. , genotype 2a, genotype 2b, genotype 2c), genotype 3 (e.g., genotype 3a), genotype 4 (e.g., genotype 4a), genotype 5, and genotype 6. In further embodiments, the HCV genotype is an HCV quasispecies. As used herein, the term "quasispecies" refers to a viral population that contains a different genetic sequence as a result of the incorporation or deletion of alternative or additional gene sequences, either correct or erroneous. Quasispecies may result, for example, from the genetic copy process of other gene sequences that are already present. Quasispecies may also arise in the context of the evolutionary process, in response to a host immune response, or in response to a therapeutic treatment. As used herein, quasispecies include both the variant species and the wild-type species from which the variant species derived.
D. Methods and Compositions for Diagnostics and Detection
[0189] In certain embodiments, any of the anti-HCV antibodies provided herein is useful for detecting the presence of HCV E2 protein or fragment thereof in a biological sample. The term "detecting" as used herein encompasses quantitative or qualitative detection. In certain embodiments, a biological sample comprises blood, serum, a cell or tissue, such as liver tissue from a liver biopsy.
[0190] In one embodiment, an anti-HCV antibody for use in a method of diagnosis or detection is provided. In a further aspect, a method of detecting the presence of HCV in a biological sample is provided. In certain embodiments, the method comprises contacting the biological sample with an anti-HCV antibody as described herein under conditions permissive for binding of the anti-HCV antibody to HCV, and detecting whether a complex is formed between the anti-HCV antibody and HCV. Such method may be an in vitro or in vivo method. In a further aspect, a method of detecting the presence of HCV E2 protein or fragment thereof in a biological sample is provided. In certain embodiments, the method comprises contacting the biological sample with an anti-HCV antibody as described herein under conditions permissive for binding of the anti-HCV antibody to HCV E2 protein, and detecting whether a complex is formed between the anti-HCV antibody and HCV E2 protein. Such method may be an in vitro or in vivo method. In one embodiment, an anti-HCV antibody is used to select subjects eligible for therapy with an anti-HCV antibody, e.g. where HCV or HCV E2 protein is a biomarker for selection of patients.
[0191] In yet a further aspect, there is provided a diagnostic test apparatus and method for determining or detecting the presence of HCV in a sample. The apparatus may comprise, as a reagent, one or more anti-HCV antibodies or fragments thereof as described herein. The antibody/ies may, for example, be immobilized on a solid support (e.g., on a microtiter assay plate, or on a particulate support) and serve to "capture" HCV particles from a sample (e.g., a blood or serum sample or other clinical specimen - such as a liver biopsy). The captured virus particles may then be detected by, for example, adding a further, labeled, reagent which binds to the captured virus particles. Conveniently, the assay may take the form of an ELISA, especially a sandwich-type ELISA, but any other assay format could in principle be adopted (e.g., radioimmunoassay, Western blot) including immunochromatographic or dipstick-type assays.
[0192] For diagnostic purposes, the anti-HCV antibodies or fragments thereof as described herein may either be labeled or unlabelled. Unlabelled antibodies can be used in combination
with other labeled antibodies (second antibodies). Alternatively, the antibodies can be directly labeled. A wide variety of labels may be employed - such as radionuclides, fluors, enzymes, enzyme substrates, enzyme cofactors, enzyme inhibitors, ligands (particularly haptens), etc. Labels include, but are not limited to, labels or moieties that are detected directly (such as fluorescent, chromophoric, electron-dense, chemiluminescent, and radioactive labels), as well as moieties, such as enzymes or ligands, that are detected indirectly, e.g., through an enzymatic reaction or molecular interaction. Exemplary labels include, but are not limited to, the radioisotopes 32 P, 14 C, 125 I, 3 H, and 131 I, fluorophores such as rare earth chelates or fluorescein and its derivatives, rhodamine and its derivatives, dansyl, umbelliferone, luceriferases, e.g., firefly luciferase and bacterial luciferase (U.S. Patent No. 4,737,456), luciferin, 2,3-dihydrophthalazinediones, horseradish peroxidase (HRP), alkaline phosphatase, β-galactosidase, glucoamylase, lysozyme, saccharide oxidases, e.g., glucose oxidase, galactose oxidase, and glucose-6-phosphate dehydrogenase, heterocyclic oxidases such as uricase and xanthine oxidase, coupled with an enzyme that employs hydrogen peroxide to oxidize a dye precursor such as HRP, lactoperoxidase, or microperoxidase, biotin/avidin, spin labels, bacteriophage labels, stable free radicals, and the like.
[0193] Since the anti-HCV antibodies or fragments thereof as described herein can bind to HCV from any of genotypes 1-6, the assay apparatus and corresponding method should be capable of detecting in a sample HCV representative from any of these genotypes or quasispecies of these genotypes.
[0194] In some embodiments, the sample is compared to a control sample. In some embodiments, the control sample is from an individual known to be infected with HCV. In some embodiments, the individual is known to infected with one or more HCV genotypes selected from the group consisting of genotype 1 {e.g., genotype la and genotype lb), genotype 2 {e.g., genotype 2a, genotype 2b, genotype 2c), genotype 3 {e.g., genotype 3a), genotype 4 {e.g., genotype 4a), genotype 5, and genotype 6. In some embodiments, the individual is known to be infected with one or more HCV quasispecies. In some
embodiments, the control sample is from an individual known not to be infected with HCV.
[0195] In some embodiments, any of the methods of treatment described are based on the determination or detection of HCV in a sample by any of the anti-HCV antibodies or fragments thereof described herein. As used herein, "based upon" includes (1) assessing, determining, or measuring the subject's characteristics as described herein (and preferably selecting a subject suitable for receiving treatment); and (2) administering the treatment(s) as described herein.
[0196] In some embodiments, a method is provided for identifying an individual suitable or not suitable (unsuitable) for treatment with the anti-HCV antibody or fragment thereof. In a further embodiment, an individual suitable for treatment is administered an anti-HCV antibody as disclosed herein.
[0197] In some embodiments, a method is providing for selecting or not selecting an individual for treatment with the anti-HCV antibody or fragment thereof, the method comprising: a) assessing the viral load and/or viral titer in a biological sample from the individual, and b) selecting the individual for treatment with an anti-HCV antibody or fragment thereof if the viral load is at least 5 IU/mL. In some embodiments, the viral load is at least 5 IU/mL, 10 IU/mL, 20 IU/mL, 40 IU/mL, 80 IU/mL, 100 IU/mL, 500 IU/mL, 1,000 IU/mL, 10,000 IU/mL, 100,000 IU/mL, 200,000 IU/mL, 300,000 IU/mL, 400,000 IU/mL, 500,000 IU/mL, 600,000 IU/mL, 700,000 IU/mL, 800,000 IU/mL, 900,000 IU/mL, or 1,000,000 IU/mL, inclusive, including any values in between these numbers.
[0198] Exemplary disorders that may be diagnosed using an antibody of the invention include acute HCV infection and chronic HCV infection.
[0199] In a further aspect, there is provided an assay method for identifying an agent that improves or enhances the efficacy of the neutralizing activity of the anti-HCV antibody or fragment thereof as described herein.
[0200] Provided herein is an assay method for identifying an agent that improves or enhances the efficacy of the neutralizing activity of the anti-HCV antibody or fragment thereof against hepatitis C virus, comprising the steps of: (a) contacting said anti-HCV antibody or antigen binding fragment thereof with an agent to be tested; and (b) determining whether the agent improves or enhances the efficacy of the anti-HCV antibody or antigen binding fragment thereof in neutralizing the infectivity of hepatitis C virus.
[0201] In some embodiments, the ability of the agent to improve or enhance the efficacy of the neutralizing activity of the anti-HCV antibody or fragment thereof against hepatitis C virus is compared to a control. In some embodiments, the control is the anti-HCV antibody or fragment thereof in the absence of the agent. In some embodiments, the control is humanized antibody or fragment thereof with a placebo, e.g., water, saline, sugar water, etc.
[0202] As used herein, the term "agent" may be a single entity or it may be a combination of entities.
[0203] The agent may be an organic compound or other chemical. The agent may be a compound, which is obtainable from or produced by any suitable source, whether natural or artificial. The agent may be an amino acid molecule, a polypeptide, or a chemical derivative
thereof, or a combination thereof. The agent may even be a polynucleotide molecule - which may be a sense or an anti-sense molecule. In some embodiments, the agent is an antibody. In some embodiments, the agent is a cytokine (such as interferon- ). In some embodiments, the agent is a direct acting antiviral agent. In further embodiments, the direct acting antiviral agent is viral protease inhibitor or a viral polymerase inhibitor. In some embodiments, the agent is an indirect acting viral agent.
[0204] The agent may be designed or obtained from a library of compounds, which may comprise peptides, as well as other compounds, such as small organic molecules.
[0205] By way of example, the agent may be a natural substance, a biological
macromolecule, or an extract made from biological materials such as bacteria, fungi, or animal (particularly mammalian) cells or tissues, an organic or an inorganic molecule, a synthetic agent, a semi- synthetic agent, a structural or functional mimetic, a peptide, a peptidomimetics, a derivatized agent, a peptide cleaved from a whole protein, or a peptides synthesized synthetically (such as, by way of example, either using a peptide synthesizer or by recombinant techniques or combinations thereof, a recombinant agent, an antibody, a natural or a non-natural agent, a fusion protein or equivalent thereof and mutants, derivatives or combinations thereof.
[0206] Typically, the agent will be an organic compound. Typically the organic compounds will comprise two or more hydrocarbyl groups. Here, the term "hydrocarbyl group" means a group comprising at least C and H and may optionally comprise one or more other suitable substituents. Examples of such substituents may include halo-, alkoxy-, nitro-, an alkyl group, a cyclic group etc. In addition to the possibility of the substituents being a cyclic group, a combination of substituents may form a cyclic group. If the hydrocarbyl group comprises more than one C then those carbons need not necessarily be linked to each other. For example, at least two of the carbons may be linked via a suitable element or group. Thus, the hydrocarbyl group may contain hetero atoms. Suitable hetero atoms will be apparent to those skilled in the art and include, for instance, sulphur, nitrogen and oxygen. For some applications, preferably the agent comprises at least one cyclic group. The cyclic group may be a polycyclic group, such as a non-fused polycyclic group. For some applications, the agent comprises at least the one of said cyclic groups linked to another hydrocarbyl group.
[0207] The agent may contain halo groups. Here, "halo" means fluoro, chloro, bromo or iodo.
[0208] The agent may contain one or more of alkyl, alkoxy, alkenyl, alkylene and alkenylene groups - which may be unbranched- or branched-chain.
E. Pharmaceutical Compositions and Formulations
[0209] Pharmaceutical compositions and formulations of an anti-HCV antibody as described herein are prepared by mixing such antibody having the desired degree of purity with one or more optional pharmaceutically acceptable carriers {Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980)), in the form of lyophilized formulations or aqueous solutions. Pharmaceutically acceptable carriers are generally nontoxic to recipients at the dosages and concentrations employed, and include, but are not limited to: buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride;
hexamethonium chloride; benzalkonium chloride; benzethonium chloride; phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol;
cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about 10 residues) polypeptides; proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, histidine, arginine, or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins; chelating agents such as EDTA; sugars such as sucrose, mannitol, trehalose or sorbitol; salt-forming counter-ions such as sodium; metal complexes {e.g. Zn-protein complexes); and/or non-ionic surfactants such as polyethylene glycol (PEG). Exemplary pharmaceutically acceptable carriers herein further include insterstitial drug dispersion agents such as soluble neutral-active hyaluronidase glycoproteins (sHASEGP), for example, human soluble PH-20 hyaluronidase glycoproteins, such as rHuPH20 (HYLENEX®, Baxter International, Inc.). Certain exemplary sHASEGPs and methods of use, including rHuPH20, are described in US Patent Publication Nos.
2005/0260186 and 2006/0104968. In one aspect, a sHASEGP is combined with one or more additional glycosaminoglycanases such as chondroitinases.
[0210] Buffers are used to control the pH in a range which optimizes the therapeutic effectiveness, especially if stability is pH dependent. Buffers are preferably present at concentrations ranging from about 50 mM to about 250 mM. Suitable buffering agents for use with the present invention include both organic and inorganic acids and salts thereof, such as citrate, phosphate, succinate, tartrate, fumarate, gluconate, oxalate, lactate, acetate. Additionally, buffers may comprise histidine and trimethylamine salts such as Tris.
[0211] Preservatives are added to retard microbial growth, and are typically present in a range from 0.2% - 1.0% (w/v). Suitable preservatives for use with the present invention
include octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride;
benzalkonium halides (e.g. , chloride, bromide, iodide), benzethonium chloride; thimerosal, phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol; cyclohexanol, 3-pentanol, and m-cresol.
[0212] Tonicity agents, sometimes known as "stabilizers" are present to adjust or maintain the tonicity of liquid in a composition. When used with large, charged biomolecules such as proteins and antibodies, they are often termed "stabilizers" because they can interact with the charged groups of the amino acid side chains, thereby lessening the potential for inter- and intra- molecular interactions. Tonicity agents can be present in any amount between 0.1% to 25% by weight, or more preferably between 1% to 5% by weight, taking into account the relative amounts of the other ingredients. Preferred tonicity agents include polyhydric sugar alcohols, preferably trihydric or higher sugar alcohols, such as glycerin, erythritol, arabitol, xylitol, sorbitol and mannitol.
[0213] Non-ionic surfactants or detergents (also known as "wetting agents") are present to help solubilize the therapeutic agent as well as to protect the therapeutic protein against agitation-induced aggregation, which also permits the formulation to be exposed to shear surface stress without causing denaturation of the active therapeutic protein or antibody. Non-ionic surfactants are present in a range of about 0.05 mg/ml to about 1.0 mg/ml, preferably about 0.07 mg/ml to about 0.2 mg/ml.
[0214] Suitable non-ionic surfactants include polysorbates (20, 40, 60, 65, 80, etc.), polyoxamers (184, 188, etc.), PLURONIC® polyols, TRITON®, polyoxyethylene sorbitan monoethers (TWEEN®-20, TWEEN®-80, etc.), lauromacrogol 400, polyoxyl 40 stearate, polyoxyethylene hydrogenated castor oil 10, 50 and 60, glycerol monostearate, sucrose fatty acid ester, methyl cellulose and carboxymethyl cellulose. Anionic detergents that can be used include sodium lauryl sulfate, dioctyle sodium sulfo succinate and dioctyl sodium sulfonate. Cationic detergents include benzalkonium chloride or benzethonium chloride.
[0215] The choice of pharmaceutical carrier, excipient or dilutent may be selected with regard to the intended route of administration and standard pharmaceutical practice.
Pharmaceutical compositions may comprise as - or in addition to - the carrier, excipient or dilutent any suitable binder(s), lubricant(s), suspending agent(s), coating agent(s) or solubilizing agent(s).
[0216] There may be different composition/formulation requirements dependent on the different delivery systems. By way of example, pharmaceutical compositions useful in the present invention may be formulated to be administered using a mini-pump or by a mucosal
route, for example, as a nasal spray or aerosol for inhalation or ingestible solution, or parenterally in which the composition is formulated by an injectable form, for delivery, by, for example, an intravenous, intramuscular or subcutaneous route. Alternatively, the formulation may be designed to be administered by a number of routes.
[0217] In some embodiments, an anti-HCV antibody formulation is a lyophilized anti-HCV antibody formulation. In another embodiment, an anti-HCV antibody formulation is an aqueous anti-HCV antibody formulation. Exemplary lyophilized antibody formulations are described in US Patent No. 6,267,958. Aqueous antibody formulations include those described in US Patent No. 6,171,586 and WO2006/044908, the latter formulations including a histidine-acetate buffer.
[0218] The formulation herein may also contain more than one active ingredients as necessary for the particular indication being treated, preferably those with complementary activities that do not adversely affect each other. In some embodiments, an active ingredient is a therapeutic agent. For example, it may be desirable to further provide a therapeutic agent such as a viral protease inhibitor, a viral polymerase inhibitor, a NS5A inhibitor, an interferon, a cyclophilin inhibitor, or an antibody. Viral protease inhibitors include NS3-4a protease inhibitors such as, but not limited to, Teleprevir, Boceprevir, Semeprevir (TMC435), BI201335, Danoprevir/r (RG7227), Vaniprevir (MK-7009), Narlaprevir (SCH 900518), Asunaprevir, GS-9256, GS-9451, ABT-450/r, ACH-1625, ACH-2684, and MK-5172. Viral polymerase inhibitors include nucleoside/nucleotide analogue inhibitors and non-nucleoside inhibitors of HCV RNA-dependent RNA polymerase. Such viral polymerase inhibitors include, but are not limited to, PSI-7977, Mercitabine, IDX184, PSI-938, INX-189,
Tegobuvir, Filibuvir, Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055. HCV NS5A inhibitors include, but are not limited to, Daclatasvir, PPI-461, GS-5885, and
GSK2336805. Exemplary cyclophilin inhibitors contemplated herein include, but are not limited to, Alisporivir and SCY-465. An interferon contemplated herein include, but are not limited to, interferon-a (such as interferon-a 2a or interferon-a 2b) and interferon-a derivatives (such as pegylated interferon-α 2a or pegylated interferon-α 2b). Antibodies that bind to other HCV proteins required by HCV to infect the cell are also contemplated. In some embodiments, the therapeutic agent is ribavirin. In any embodiments herein, a therapeutic agent as described herein can be used in a formulation with an anti-HCV antibody as described herein including anti-HCV antibodies described in Owsianka et al., J. Gen.
Virol, (2001), 82(Pt 8): 1877-83; WO2006/ 100449; and WO2009/081285. Such active
ingredients (such as therapeutic agents) are suitably present in combination in amounts that are effective for the purpose intended.
[0219] Active ingredients may be entrapped in microcapsules prepared, for example, by coacervation techniques or by interfacial polymerization, for example,
hydroxymethylcellulose or gelatin-microcapsules and poly-(methylmethacylate)
microcapsules, respectively, in colloidal drug delivery systems (for example, liposomes, albumin microspheres, microemulsions, nano-particles and nanocapsules) or in
macroemulsions. Such techniques are disclosed in Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980).
[0220] Stability of the proteins and antibodies described herein may be enhanced through the use of non-toxic "water-soluble polyvalent metal salts". Examples include Ca2+, Mg2+, Zn2+, Fe2+, Fe3+, Cu2+, Sn2+, Sn4+, Al2+ and Al3+. Exemplary anions that can form water soluble salts with the above polyvalent metal cations include those formed from inorganic acids and/or organic acids. Such water-soluble salts have are soluble in water (at 20°C) to at least about 20 mg/ml, alternatively at least about 100 mg/ml, alternatively at least about 200 mg/ml.
[0221] Suitable inorganic acids that can be used to form the "water soluble polyvalent metal salts" include hydrochloric, acetic, sulfuric, nitric, thiocyanic and phosphoric acid. Suitable organic acids that can be used include aliphatic carboxylic acid and aromatic acids. Aliphatic acids within this definition may be defined as saturated or unsaturated C2-9 carboxylic acids {e.g., aliphatic mono-, di- and tri-carboxylic acids). For example, exemplary monocarboxylic acids within this definition include the saturated C2-9 monocarboxylic acids acetic, proprionic, butyric, valeric, caproic, enanthic, caprylic pelargonic and capryonic, and the unsaturated C2-9 monocarboxylic acids acrylic, propriolic methacrylic, crotonic and isocro tonic acids. Exemplary dicarboxylic acids include the saturated C2-9 dicarboxylic acids malonic, succinic, glutaric, adipic and pimelic, while unsaturated C2-9 dicarboxylic acids include maleic, fumaric, citraconic and mesaconic acids. Exemplary tricarboxylic acids include the saturated C2-9 tricarboxylic acids tricarballylic and 1,2,3-butanetricarboxylic acid. Additionally, the carboxylic acids of this definition may also contain one or two hydroxyl groups to form hydroxy carboxylic acids. Exemplary hydroxy carboxylic acids include glycolic, lactic, glyceric, tartronic, malic, tartaric and citric acid. Aromatic acids within this definition include benzoic and salicylic acid.
[0222] Commonly employed water soluble polyvalent metal salts which may be used to help stabilize the encapsulated polypeptides of this invention include, for example: (1) the
inorganic acid metal salts of halides (e.g., zinc chloride, calcium chloride), sulfates, nitrates, phosphates and thiocyanates; (2) the aliphatic carboxylic acid metal salts (e.g., calcium acetate, zinc acetate, calcium proprionate, zinc glycolate, calcium lactate, zinc lactate and zinc tartrate); and (3) the aromatic carboxylic acid metal salts of benzoates (e.g., zinc benzoate) and salicylates.
[0223] Pharmaceutical formulations of anti-HCV antibodies can be designed to
immediately release an anti-HCV antibody ("immediate-release" formulations), to gradually release the antibodies over an extended period of time ("sustained-release," "controlled- release," or "extended-release" formulations), or with alternative release profiles. The additional materials used to prepare a pharmaceutical formulation can vary depending on the therapeutic form of the formulation (e.g., whether the system is designed for immediate- release or sustained-, controlled-, or extended-release). In certain variations, a sustained- release formulation can further comprise an immediate-release component to quickly deliver a priming dose following drug delivery, as well as a sustained-release component. Thus, sustained-release formulations can be combined with immediate-release formulations to provide a rapid "burst" of drug into the system as well as a longer, gradual release. For example, a core sustained-release formulation may be coated with a highly soluble layer incorporating the drug. Alternatively, a sustained-release formulation and an immediate- release formulation may be included as alternate layers in a tablet or as separate granule types in a capsule. Other combinations of different types of drug formulations can be used to achieve the desired therapeutic plasma profile.
[0224] Suitable examples of sustained-release preparations include semi-permeable matrices of solid hydrophobic polymers containing the antagonist, which matrices are in the form of shaped articles, e.g., films, or microcapsules. Examples of sustained-release matrices include polyesters, hydrogels (for example, poly(2-hydroxyethyl-methacrylate), or poly(vinylalcohol)), polylactides (U.S. Pat. No. 3,773,919), copolymers of L-glutamic acid and ethyl-L-glutamate, non-degradable ethylene-vinyl acetate, degradable lactic acid-glycolic acid copolymers such as the LUPRON DEPOT™ (injectable microspheres composed of lactic acid-glycolic acid copolymer and leuprolide acetate), and poly-D-(-)-3-hydroxybutyric acid.
[0225] The formulations to be used for in vivo administration are generally sterile. Sterility may be readily accomplished, e.g., by filtration through sterile filtration membranes.
[0226] The pharmaceutical compositions may be used in any of the methods described herein.
[0227] The pharmaceutical composition may be used among those subjects (e.g., humans) susceptible to infection with HCV i.e. to prevent or reduce/decrease the onset of HCV infection.
[0228] The pharmaceutical composition may be used among those subjects (e.g., humans) already infected with HCV i.e. to treat HCV infection. Such treatment may facilitate clearance of the virus from those subjects who are acutely or chronically infected including infected patients undergoing liver transplantation. In some embodiments, the pharmaceutical composition is used to prevent liver transplant re-infection. In some embodiments, the pharmaceutical composition is used to prevent HCV infection of the transplanted liver. In some embodiments, the pharmaceutical composition is used to prevent infection of a patient receiving an HCV infected liver transplant. In some embodiments, the pharmaceutical composition is administered by an individual before receiving a liver transplant. In some embodiments, the pharmaceutical composition is administered by an individual during receipt of a liver transplant. In some embodiments, the pharmaceutical composition is administered by an individual after receiving a liver transplant.
[0229] Thus, in a further aspect the invention provides a method for the treatment and/or prevention of hepatitis C virus infection, comprising the use of the anti-HCV antibody or the anti-HCV antibody fragment or the pharmaceutical composition. Suitably, an effective amount of the anti-HCV antibody or antibody fragment thereof or the pharmaceutical composition is administered to the subject. In some embodiments, the anti-HCV antibody or anti-HCV antibody fragment is administered in a therapeutic effective amount to effect beneficial clinical results, including, but not limited to ameliorating one or more symptoms of HCV infections or aspects of HCV infection. In some embodiments, the anti-HCV antibody or anti-HCV antibody fragment is administered in a therapeutic effective amount to reduce viral titer and/or viral load of HCV. In some embodiments, the anti-HCV antibody or anti- HCV antibody fragment is administered in a therapeutic effective amount to achieve a sustained virologic response. As used herein, the term "sustained virologic response" refers to the absence of detectable viremia twelve weeks after stopping anti-HCV treatment.
[0230] There is also provided an anti-HCV antibody or a fragment thereof or the pharmaceutical composition for use in the treatment and/or prevention of hepatitis C virus infection in a subject.
[0231] There is also provided the use of an antibody of a fragment thereof or the pharmaceutical composition in the manufacture of a composition for the treatment and/or prevention of hepatitis C virus infection in a subject.
[0232] The antibody/ies may be administered, for example, in the form of immune serum or may more preferably be a purified recombinant or monoclonal antibody. Methods of producing sera or monoclonal antibodies with the desired specificity are routine and well- known to those skilled in the art. One skilled in the art understands that the antibody/ies can be administered by various routes including, for example, injection, intubation, via a suppository, orally or topically, the latter of which can be passive, for example, by direct application of an ointment or powder containing the antibodies, or active, for example, using a nasal spray or inhalant. The antibodies can also be administered as a topical spray, if desirable, in which case one component of the composition is an appropriate propellant.
[0233] The anti-HCV antibodies and fragments thereof described herein can be
administered to a subject in accord with known methods, such as by intravenous
administration, e.g., as a bolus or by continuous infusion over a period of time, by
subcutaneous, intramuscular, intraperitoneal, intracerobrospinal, intra-articular, intrasynovial, intrathecal, or inhalation routes, generally by intravenous or subcutaneous administration.
[0234] Suitably, a passive immunization regime may conveniently comprise administration of the anti-HCV antibody of fragment thereof as described herein and/or administration of antibody in combination with other antiviral therapeutic compounds. Recently such passive immunization techniques have been used safely to treat HIV infection (Armbruster et al, J. Antimicrob. Chemother. 54, 915-920 (2004); Stiegler & Katinger, J. Antimicrob. Chemother. 51, 757-759 (2003)).
[0235] The active or passive immunization methods of the invention should allow for the protection or treatment of individuals against infection with viruses of any of genotypes 1-6 of HCV.
[0236] In some embodiments, the anti-HCV antibody or fragment thereof can be administered in combination with a second therapeutic agent (such as a viral protease inhibitor). In some embodiments, the second therapeutic agent is an antiviral therapeutic agent. In some embodiments, the anti-HCV antibody or fragment thereof is administered in combination with, sequential to, concurrently with, consecutively with, rotationally with, or intermittently with a second therapeutic agent (such as a viral protease inhibitor). In some embodiments, the administration of the combination of an anti-HCV antibody or fragment thereof and a second therapeutic agent (such as a viral protease inhibitor) ameliorates one or more symptom of HCV, reduces and/or suppresses viral titer and/or viral load, and/or prevents HCV, and/or achieves a sustained virologic response more than treatment with the anti-HCV antibody or fragment thereof or second therapeutic agent alone. In some
embodiments, the anti-HCV antibody or fragment thereof and the second therapeutic agent are provided in separate dosage forms. In some embodiments, the anti-HCV antibody or fragment thereof and the second therapeutic agent are provided in the same dosage form.
F. Therapeutic Methods of Use
[0237] The invention provides methods for treating or preventing an HCV infection in a subject comprising administering to the subject an effective amount of an anti-HCV antibody described herein. In some embodiments, the subject is human. In some embodiments, the subject has been diagnosed with HCV infection. Any of the anti-HCV antibodies provided herein may be used in therapeutic methods.
[0238] The anti-HCV antibodies and fragments thereof or a pharmaceutical composition comprising same are useful in reducing, eliminating, or inhibiting HCV infection and can be used for treating any pathological condition that is characterized, at least in part, by HCV infection. The anti-HCV antibodies and fragments thereof and/or the pharmaceutical composition can be used for treating an HCV infection. The anti-HCV antibodies and fragments thereof and/or the pharmaceutical composition can also be used in methods for preventing a HCV infection.
[0239] The term "hepatitis C virus" or "HCV" is well understood in the art and refers to a virus which is a member of the genus Hepacivirus of the family flaviviridae. HCV is a lipid enveloped virus having a diameter of approximately 55-65 nm in diameter with a positive strand RNA genome. The hepatitis C virus species is classified into six genotypes (1-6) with several subtypes within each genotype. In some embodiments, the subject is infected with one or more HCV genotypes selected from the group consisting of genotype 1 (e.g., genotype la and genotype lb), genotype 2 (e.g., genotype 2a, genotype 2b, genotype 2c), genotype 3 (e.g., genotype 3a), genotype 4 (e.g., genotype 4a), genotype 5, and genotype 6. In North America, genotype la predominates followed by lb, 2a, 2b, and 3a. In Europe, genotype lb is predominant followed by 2a, 2b, 2c, and 3a. Genotypes 4 and 5 are found almost exclusively in Africa. Genotype 6 is found in Southeast Asia and Australia. In some embodiments, an anti-HCV antibody or fragment thereof as described herein are useful in methods of treating an individual infected with an HCV genotype attributed to a specific geographical population (such as genotype 2 in Japan). See Simmonds P., J Gen Virol., (2004)., 85(Pt 11):3173-88 for non-limiting examples of specific geographical populations that can be treated with the anti-HCV antibodies described herein.
[0240] Provided herein are methods for treating a hepatitis C virus infection in a subject, comprising administering an effective amount of the anti-HCV antibody fragment thereof described herein. In some embodiments, the anti-HCV antibodies and fragments thereof described herein are useful in methods of treating an acute HCV infection. In some embodiments, treating an acute HCV infection includes reducing, eliminating, or inhibiting an acute HCV infection. The term "acute hepatitis C virus infection" or "acute HCV infection," as used herein, refers to the first 6 months after infection with HCV. In some embodiments, a subject with an acute HCV infection will not develop any symptoms (i.e., free of acute HCV infection symptoms). Between 60% to 70% of subjects with acute HCV infection develop no symptoms during the acute phase. In some embodiments, a subject with acute HCV infection will develop symptoms. In some embodiments, the methods of treatment described herein ameliorate (e.g. , reduce incidence of, reduce duration of, reduce or lessen severity of) of one or more symptoms of acute HCV infection. In the minority of patients who experience acute phase symptoms, the symptoms are generally mild and nonspecific, and rarely lead to a specific diagnosis of hepatitis C. Symptoms of acute hepatitis C infection include decreased appetite, fatigue, abdominal pain, jaundice, itching, and flu-like symptoms. In some embodiments, the subject with acute HCV infection is infected with HCV of the genotype 1. Treatment during the acute HCV injection of genotype 1 has a greater than 90% success rate with half the treatment time required for chronic infections.
[0241] In some embodiments, the anti-HCV antibodies and fragments thereof described are useful in methods of treating a chronic HCV infection. In some embodiments, treating a chronic HCV infection includes reducing, eliminating, or inhibiting a chronic HCV infection. The term "chronic hepatitis C virus infection" or "chronic HCV infection," as used herein, refers to an infection with HCV which persisting for more than six months. In some embodiments, the methods of treatment described herein ameliorate (e.g. , reduce incidence of, reduce duration of, reduce or lessen severity of) of one or more symptoms of chronic HCV infection. Symptoms of chronic HCV infection include fatigue, marked weight loss, flu-like symptoms, muscle pain, joint pain, intermittent low-grade fevers, itching, sleep disturbances, abdominal pain (especially in the right upper quadrant), appetite changes, nausea, diarrhea, dyspepsia, cognitive changes, depression, headaches, and mood swings. Once chronic HCV has progressed to cirrhosis, signs and symptoms may appear that are generally caused by either decreased liver function or increased pressure in the liver circulation, a condition known as portal hypertension. Possible signs and symptoms of liver
cirrhosis include ascites, bruising and bleeding tendency, bone pain, varices (especially in the stomach and esophagus), fatty stools (steatorrhea), jaundice, and a syndrome of cognitive impairment known as hepatic encephalopathy. In some embodiments, the chronic HCV infection may result in hepatocellular carcinoma. Chronic HCV infection can be further divided into two types (either or both of which are included in the methods of treatment provided herein) chronic active HCV infection and chronic persistent HCV infection. Chronic active HCV infection is HCV which is cause active damage to the liver. Chronic persistent HCV infection is a chronic HCV infection which is not currently causing damage to the liver, although pre-existing liver damage may be present.
[0242] The term "HCV-associated diseases" or "HCV-associated disorders" as used herein, refers to an infection with HCV or a disease or disorder that is associated with HCV infection such as liver disease. Accordingly, in some embodiments, an anti-HCV antibody or fragment as described herein prevents development of an HCV-associated disease. In some
embodiments, the HCV-associated disease is HCV infection. In some embodiments, the HCV-associated disease is a liver disease such as, but not limited to, cirrhosis or
hepatocellular carcinoma. In some embodiments, an anti-HCV antibody or fragment as described herein is used in methods of treating an HCV-associated disease. In some embodiments, the HCV-associated disease is HCV infection. In some embodiments, the HCV-associated disease is a liver disease such as, but not limited to, cirrhosis or
hepatocellular carcinoma.
[0243] In some embodiments, the anti-HCV antibodies or fragments thereof may be administered to the subject infected with HCV prior to, concurrent with, or subsequent to a liver transplant. In some embodiments, an anti-HCV antibody or fragment thereof prevents liver transplant re-infection. In some embodiments, an anti-HCV antibody or fragment thereof prevents HCV infection of the transplanted liver.
[0244] In some embodiments of any of the methods of treating, the anti-HCV antibodies and fragments thereof described herein are useful in methods of treatment including suppressing one or more aspects of a HCV infection. In some embodiments, the HCV infection is a chronic HCV infection. In some embodiments, the HCV infection is an acute HCV infection. In some embodiments, the methods described herein suppress a HCV- associated laboratory finding (e.g., ALAT, AST, and GGTP levels in blood), viral replication, viral titer, viral load, or viremia.
[0245] In some embodiments, the methods described herein suppress or reduce viral titer. "Viral titer" is known in the art and indicates the amount of virus in a given biological
sample. In some embodiments, the methods described herein suppress or reduce viremia. "Viremia" is known in the art as the presence of virus in the bloodstream and/or viral titer in a blood or serum sample. In some embodiments, the methods described herein suppress or reduce viral load. "Viral load" refers to the amount of hepatitis C virus in a person's blood. The results of a hepatitis C viral load test (known as a viral RNA test or HCV RNA test) are usually expressed as International Units/mL (IU/mL) or RNA copies/mL. A subject with a hepatitis C viral load of 1 million IU/mL or more is considered to have a high viral load. Amount of virus (e.g., viral titer or viral load) are indicated by various measurements, including, but not limited to amount of viral nucleic acid, the presence of viral particles, replicating units (RU), plaque forming units (PFU). Amount of virus such as high viral load, low viral load or undetectable viral load can be defined according to a clinical acceptable parameter established by those skilled in the art. In some embodiments, an undetectable viral load is defined by the limit of the assay for detecting HCV. Generally, for fluid samples such as blood and urine, amount of virus is determined per unit fluid, such as milliliters. For solid samples, such as tissue samples, amount of virus is determined per weight unit, such as grams. Methods for determining amount of virus are known in the art and are also described herein. In some embodiments, the methods described herein result in a sustained virologic response for at least 12 weeks after stopping the treatment. In some embodiments, the methods described herein reduce serum alanine aminotransferase (ALT) levels.
[0246] In some embodiments, the subject treated with the anti-HCV antibodies and fragments thereof described herein is at risk for rapid HCV infection progression. Factors that have been reported to influence the rate of HCV disease progression include age (increasing age associated with more rapid progression), gender (males have more rapid disease progression than females), alcohol consumption (associated with an increased rate of disease progression), HIV co-infection (associated with a markedly increased rate of disease progression), and fatty liver (the presence of fat in liver cells has been associated with an increased rate of disease progression).
[0247] The anti-HCV antibodies and fragments thereof described herein can also be used in methods for preventing a HCV infection. In some embodiments, the anti-HCV antibodies and fragments thereof described herein are useful in methods of preventing an acute HCV infection. In some embodiments, the anti-HCV antibodies and fragments thereof described herein are useful in methods of preventing a chronic HCV infection. In some embodiments, the anti-HCV antibodies and fragments can be used in methods for preventing a HCV infection in a subject susceptible to infection with HCV. In some embodiments, the anti-HCV
antibodies and fragments thereof can also be used in methods for preventing a HCV infection in a subject exposed to or potentially exposed to HCV. "Exposure" to HCV denotes an encounter or potential encounter with HCV which could result in an HCV infection.
Generally, an exposed subject is a subject that has been exposed to HCV by a route by which HCV can be transmitted. In some embodiments, the subject has been exposed to or potentially exposed to blood of a subject with an HCV infection or blood from a subject which may or may not be infected with HCV (i.e. , HCV infection status of the blood exposure is unknown). HCV is often transmitted by blood-to-blood contact. In some embodiments, the subject has been exposed to or potentially exposed to HCV by, but not limited to, use of blood products (e.g., a blood transfusion), "needle stick" accidents, sharing drug needles, snorting drugs, a sexual partner, iatrogenic medical or dental exposure, needles used in body piercings and tattoos, or a child whose mother has an HCV infection. In some embodiments of the methods of prevention, the anti-HCV antibodies and fragments thereof described herein will be administered at the time or within any of about one day, one week, or one month of the exposure or potential exposure to HCV.
[0248] In some embodiments of any of the methods described herein, the subject is a human or chimpanzee. In some embodiments, the subject is a human.
[0249] In some embodiments, provided herein is a method for treating or preventing a HCV infection in a subject comprising administering an anti-HCV antibody or fragment (such as an antibody described herein including V361 and V362, and any other antibody that binds HCV E2 protein) in combination with a second therapeutic agent. In some
embodiments, the second therapeutic agent is an antiviral therapeutic agent. In some embodiments, the second therapeutic agent is selected from the group consisting of: a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a second anti-HCV antibody, and a combination thereof. In some embodiments, the second therapeutic agent is a viral protease inhibitor selected from the group consisting of Teleprevir,
Boceprevir, Semeprevir (TMC435), BI201335, Danoprevir/r (RG7227), Vaniprevir (MK- 7009), Narlaprevir (SCH 900518), Asunaprevir, GS-9256, GS-9451, ABT-450/r, ACH- 1625, ACH-2684, and MK-5172. In some embodiments, the second therapeutic agent is a viral polymerase inhibitor selected from the group consisting of PSI-7977, Mercitabine, IDX184, PSI-938, INX- 189, Tegobuvir, Filibuvir, Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055. In some embodiments, the second therapeutic agent is a HCV NS5A inhibitor selected from the group consisting of: Daclatasvir, PPI-461, GS-5885, and GSK2336805. In some embodiments, the second therapeutic agent is a cyclophilin inhibitor selected from the
group consisting of: Alisporivir and SCY-465. In some embodiments, the second therapeutic agent is pegylated interferon-a 2a or pegylated interferon-a 2b. In some embodiments, the second therapeutic agent is an antibody as described herein. In some embodiments, the second therapeutic agent is ribavirin. In some embodiments, the method comprises administering the anti-HCV antibody or fragment thereof in combination with, sequential to, concurrently with, consecutively with, rotationally with, or intermittently with a second therapeutic agent. In some embodiment, the method comprising administering the combination of an anti-HCV antibody or fragment thereof and a second therapeutic agent ameliorates one or more symptom of HCV, reduces and/or suppresses viral titer and/or viral load, and/or prevents HCV, and/or achieves a sustained virologic response for at least twelve weeks after stopping treatment more than treatment with the ant-HCV antibody or fragment thereof or second therapeutic agent alone. In some embodiments, the viral in the subject has been reduced to an undetectable level after treatment. In some embodiments in any of the methods described herein, the treatment is an interferon-free treatment.
[0250] In some embodiments of any of the methods described herein, the subject is not responsive to interferon treatment. In some embodiments, the method comprises
administering an anti-HCV antibody or fragment thereof to a subject that is not responsive to interferon treatment. In some embodiments, the method comprises administering an anti- HCV antibody or fragment thereof and an interferon to a subject that is not responsive to interferon treatment alone. As described herein, subjects that not responsive to interferon treatment include subjects that have detectable HCV during the course of treatment with interferon.
[0251] Also provided herein is a method of preventing developing resistance to treatment, comprising administering an anti-HCV antibody or fragment (such as an antibody described herein including V361 and V362, and any other antibody that binds HCV E2 protein) and a second therapeutic agent. In some embodiments, the second therapeutic agent is an antiviral therapeutic agent. In some embodiments, the second therapeutic agent is selected from the group consisting of: a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a second anti-HCV antibody, and a combination thereof. In some embodiments, the second therapeutic agent is a viral protease inhibitor selected from the group consisting of Teleprevir, Boceprevir, Semeprevir (TMC435), BI201335, Danoprevir/r (RG7227), Vaniprevir (MK-7009), Narlaprevir (SCH 900518), Asunaprevir, GS-9256, GS- 9451, ABT-450/r, ACH-1625, ACH-2684, and MK-5172. In some embodiments, the second therapeutic agent is a viral polymerase inhibitor selected from the group consisting of PSI-
7977, Mercitabine, IDX184, PSI-938, INX-189, Tegobuvir, Filibuvir, Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055. In some embodiments, the second therapeutic agent is a HCV NS5A inhibitor selected from the group consisting of: Daclatasvir, PPI-461, GS- 5885, and GSK2336805. In some embodiments, the second therapeutic agent is a cyclophilin inhibitor selected from the group consisting of: Alisporivir and SCY-465. In some embodiments, the second therapeutic agent is pegylated interferon-a 2a or pegylated interferon-a 2b. In some embodiments, the second therapeutic agent is an antibody as described herein. In some embodiments, the second therapeutic agent is ribavirin. In some embodiments, the method comprises administering the anti-HCV antibody or fragment thereof in combination with, sequential to, concurrently with, consecutively with, rotationally with, or intermittently with a second therapeutic agent. In some embodiments, the viral resistance in the subject is undetectable or low. Methods of determining undetectable or low viral resistance are known in the art. For example, HCV RNA extracted from a biological sample collected every day up to 12 weeks post treatment from an individual administered an anti-HCV antibody a described herein is sequenced and compared to a control sequence (such as a wild-type sequence) to determine if mutations have arisen in the HCV that is infecting the individual. No mutations or low copy numbers of mutations as compared to the whole sample are indicative that viral resistance in the subject receiving treatment with an anti-HCV antibody is undetectable or low. In some embodiments, the viral resistance in a subject receiving an anti-HCV antibody and a second therapeutic agent is undetectable or low. In some embodiments, the viral resistance in a subject receiving an anti-HCV antibody and a second therapeutic agent is undetectable or low as compared to a subject receiving a second therapeutic agent alone. In some embodiments, the viral resistance in a subject receiving an anti-HCV antibody and a second therapeutic agent is undetectable or low as compared to a subject receiving an anti-HCV antibody alone.
[0252] In a further aspect, the invention provides for the use of an anti-HCV antibody in the manufacture or preparation of a medicament. In one embodiment, the medicament is for treatment of HCV infection. In a further embodiment, a medicament comprising an anti- HCV antibody for use in a method of treating HCV infection comprises administering to an individual having an HCV infection an effective amount of the medicament comprising an anti-HCV antibody. In one such embodiment, the method further comprises administering to the individual an effective amount of at least one additional therapeutic agent. In some embodiments, the invention provides for the use of an anti-HCV antibody as described herein (such as MRCT10.1 or V362) in combination with a second therapeutic agent in the
manufacture or preparation of a medicament. In some embodiments, the second therapeutic agent is an antiviral therapeutic agent. In some embodiments, the second therapeutic agent is selected from the group consisting of: a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a second anti-HCV antibody, and a combination thereof. In some embodiments, the second therapeutic agent is a viral protease inhibitor selected from the group consisting of Teleprevir, Boceprevir, Semeprevir (TMC435), BI201335,
Danoprevir/r (RG7227), Vaniprevir (MK-7009), Narlaprevir (SCH 900518), Asunaprevir, GS-9256, GS-9451, ABT-450/r, ACH-1625, ACH-2684, and MK-5172. In some
embodiments, the second therapeutic agent is a viral polymerase inhibitor selected from the group consisting of PSI-7977, Mercitabine, IDX184, PSI-938, INX-189, Tegobuvir,
Filibuvir, Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055. In some
embodiments, the second therapeutic agent is a HCV NS5A inhibitor selected from the group consisting of: Daclatasvir, PPI-461, GS-5885, and GSK2336805. In some embodiments, the second therapeutic agent is a cyclophilin inhibitor selected from the group consisting of: Alisporivir and SCY-465. In some embodiments, the second therapeutic agent is pegylated interferon-a 2a or pegylated interferon-a 2b. In some embodiments, the second therapeutic agent is an antibody as described herein. In some embodiments, the second therapeutic agent is ribavirin. Such combination therapies noted above encompass combined administration (where two or more therapeutic agents are included in the same or separate formulations), and separate administration, in which case, administration of the antibody of the invention can occur prior to, simultaneously, and/or following, administration of the additional therapeutic agent and/or adjuvant.
[0253] In a further aspect, the invention provides pharmaceutical formulations as described herein comprising any of the anti-HCV antibodies provided herein, e.g., for use in any of the above therapeutic methods.
[0254] An anti-HCV antibody of the invention (and any additional therapeutic agent) can be administered by any suitable means, including parenteral, intrapulmonary, and intranasal, and, if desired for local treatment, intralesional administration. Parenteral infusions include intramuscular, intravenous, intraarterial, intraperitoneal, or subcutaneous administration. Dosing can be by any suitable route, e.g. by injections, such as intravenous or subcutaneous injections, depending in part on whether the administration is brief or chronic. Various dosing schedules including but not limited to single or multiple administrations over various time- points, bolus administration, and pulse infusion are contemplated herein.
[0255] Anti-HCV antibodies of the invention would be formulated, dosed, and administered in a fashion consistent with good medical practice. Factors for consideration in this context include the particular disorder being treated, the particular mammal being treated, the clinical condition of the individual patient, the cause of the disorder, the site of delivery of the agent, the method of administration, the scheduling of administration, and other factors known to medical practitioners. The antibody need not be, but is optionally formulated with one or more agents currently used to prevent or treat the disorder in question. The effective amount of such other agents depends on the amount of antibody present in the formulation, the type of disorder or treatment, and other factors discussed above. These are generally used in the same dosages and with administration routes as described herein, or about from 1 to 99% of the dosages described herein, or in any dosage and by any route that is
empirically/clinically determined to be appropriate.
[0256] For the prevention or treatment of disease, the appropriate dosage of an antibody of the invention (when used alone or in combination with one or more other additional therapeutic agents) will depend on the type of disease to be treated, the type of antibody, the severity and course of the disease, whether the antibody is administered for preventive or therapeutic purposes, previous therapy, the patient's clinical history and response to the antibody, and the discretion of the attending physician. The antibody is suitably administered to the patient at one time or over a series of treatments. Depending on the type and severity of the disease, about 1 μg/kg to 15 mg/kg (e.g. O. lmg/kg-lOmg/kg) of antibody can be an initial candidate dosage for administration to the patient, whether, for example, by one or more separate administrations, or by continuous infusion. One typical daily dosage might range from about 1 μg/kg to 100 mg/kg or more, depending on the factors mentioned above. For repeated administrations over several days or longer, depending on the condition, the treatment would generally be sustained until a desired suppression of disease symptoms occurs. One exemplary dosage of the antibody would be in the range from about 0.05 mg/kg to about 10 mg/kg. Thus, one or more doses of about 0.5 mg/kg, 2.0 mg/kg, 4.0 mg/kg or 10 mg/kg (or any combination thereof) may be administered to the patient. Such doses may be administered intermittently, e.g. every week or every three weeks (e.g. such that the patient receives from about two to about twenty, or e.g. about six doses of the antibody). An initial higher loading dose, followed by one or more lower doses may be administered. An exemplary dosing regimen comprises administering a monthly subcutaneous dose of 1 to 2 mg/kg anti-HCV antibody to an individual for up to six months. In some embodiments, the individual has chronic HCV. Another exemplary dosing regimen comprises administering a
dose (such as an intravenous dose) of an anti-HCV antibody as described herein to an individual receiving a liver transplant. In some embodiments, the individual is administered a dose (such as an intravenous dose) of an anti-HCV antibody before receiving a liver transplant. In some embodiments, the individual is administered a dose (such as an intravenous dose) of an anti-HCV antibody while receiving a liver transplant. In some embodiments, the individual is administered a dose (such as an intravenous dose) of an anti- HCV antibody after receiving a liver transplant. In some embodiments, the individual is administered a dose (such as an intravenous dose) of an anti-HCV antibody for at least one month after receiving a liver transplant. However, other dosage regimens may be useful. The progress of this therapy is easily monitored by conventional techniques and assays. In some embodiments, the progress of the therapy is monitored by a reduction or suppression in viral load. In some embodiments, the progress of the therapy is monitored by a reduction of ALT levels.
[0257] Any anti-HCV antibodies described herein, including anti-HCV antibodies described in Owsianka et al., J. Gen. Virol, (2001), 82(Pt 8): 1877-83; WO2006/ 100449; and WO2009/081285, can be used in any of the embodiments of any of the methods described herein. In some embodiments of any of the methods described herein, an anti-HCV antibody or fragment thereof comprises a variable heavy chain region selected from the group consisting of SEQ ID NOs: 17, 18, 19, 20, 21, 22, and 23, and a variable light chain region selected from the group consisting of SEQ ID NOs:24, 25, and 26. In some embodiments of any of the methods described herein, the fragment of the anti-HCV antibody is selected from the group consisting of a Fab fragment, a Fab' fragment, a F(ab')2 fragment, a scFv, a Fv, and a diabody. In some embodiments of any of the methods described herein, the anti-HCV antibody or fragment thereof binds to HCV. In some embodiments of any of the methods described herein, the anti-HCV antibody or fragment thereof is capable of binding to HCV E2 protein, soluble HCV E2 protein, or a heterodimer of HCV El protein and HCV E2 protein. In some embodiments of any of the methods described herein, the anti-HCV antibody or fragment thereof binds HCV E2 protein. In some embodiments of any of the methods described herein, the HCV E2 protein is from one or more of the HCV genotypes selected from the group consisting of genotype 1 {e.g., genotype la and genotype lb), genotype 2 {e.g., genotype 2a, genotype 2b, genotype 2c), genotype 3 {e.g., genotype 3a), genotype 4 {e.g., genotype 4a), genotype 5, and genotype 6. In some embodiments of any of the methods described herein, the anti-HCV antibody or fragment thereof inhibits the interaction of HCV E2 protein with CD81. In some embodiments of any of the methods
described herein, the anti-HCV antibody or fragment thereof prevents and/or inhibits HCV entry into the cell. In some embodiments, the cell is a liver cell, e.g., hepatocyte.
G. Articles of Manufacture or Kits
[0258] In another aspect of the invention, an article of manufacture or kit containing anti- HCV antibodies or fragments thereof useful for the treatment, prevention and/or diagnosis of the disorders described above is provided. The article of manufacture or kit comprises a container and a label or package insert on or associated with the container. Suitable containers include, for example, bottles, vials, syringes, IV solution bags, etc. The containers may be formed from a variety of materials such as glass or plastic. The container holds a composition which is by itself or combined with another composition effective for treating, preventing and/or diagnosing the condition and may have a sterile access port (for example the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle). At least one active agent in the composition is an anti-HCV antibody or fragment thereof of the invention. The label or package insert indicates that the composition is used for treating the condition of choice. Moreover, the article of manufacture or kit may comprise (a) a first container with a composition contained therein, wherein the composition comprises an anti-HCV antibody or fragment thereof of the invention; and (b) a second container with a composition contained therein, wherein the composition comprises a further cytotoxic or otherwise therapeutic agent. The article of manufacture or kit in this embodiment of the invention may further comprise a package insert indicating that the compositions can be used to treat a particular condition. Alternatively, or additionally, the article of manufacture or kit may further comprise a second (or third) container comprising a pharmaceutically-acceptable buffer, such as bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's solution and dextrose solution. It may further include other materials desirable from a commercial and user standpoint, including other buffers, diluents, filters, needles, and syringes.
[0259] The invention also provides diagnostic kits, for example, research, detection and/or diagnostic kits. Such kits typically contain the anti-HCV antibody or fragment thereof as described herein. Suitably, the antibody is labeled or a secondary labeling reagent is included in the kit. Preferably, the kit is labeled with instructions for performing the intended application, for example, for performing an in vivo imaging assay.
[0260] Although the foregoing invention has been described in some detail by way of illustration and example for purposes of clarity of understanding, the descriptions and examples should not be construed as limiting the scope of the invention.
EXAMPLES
Example 1: Generation of anti-HCV antibodies
[0261] To identify specific residues to alter for generation of anti-HCV antibody variants, combinatorial antibody phage libraries against soluble genotype la HCV E2 protein, which contained the first 661 residues of the extracellular domain (sE2661), were used for affinity maturation of MRCT10.1 antibody. The MRCT10.1 antibody has the HVR-H1, HVR-H2, and HVR-H3 amino acid sequences found in SEQ ID NO: 13 and the HVR-L1, HVR-L2, and HVR-L3 amino acid sequences found in SEQ ID NO: 19 as disclosed in WO2009/081285, which is incorporated herein by reference in its entirety.
MRCT 10.1.1 heavy chain amino acid sequence
EVQLQESGPGLVKPSETLSLTCTVSGDSITSGYWNWIRQPPGRALEWMGYISYSGST
YYNLSLRSRITISRDTSKNQYSLRLSSVTAADTAMYYCALITTTTYAMDYWGQGTTV
TVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFP
AVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPC
PAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHN
AKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQ
PREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLD
SDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG (SEQ ID
NO:29)
MRCT 10.1.1 light chain amino acid sequence
DIVLTQSPSSLSASVGDRVTITCRASESVDGYGNSFLHWFQQKPGKAPKLLIYLASNL NSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQNNVDPWTFGQGTKLEIKRTVAA PSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSK DSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO:31)
[0262] Libraries were synthesized by site-directed mutagenesis following a protocol adapted from Sidhu et al. Phage Display: A Practical Approach, (2004), pp. 27-41, Oxford University Press. Briefly, variable domains of MRCT10.1.1 were amplified by PCR using oligonucleotides: 5'-
GCTACAAATGCCTATGC AGATATCGTGTTG ACCCAGTCCCCGAGC-3 ' (SEQ ID NO:37); 5'-
CCGTGG ACGTTCGG AC AGGGT ACC A AGTTGG AG ATC A A ACG A ACTGTG- 3 ' (SEQ ID NO:38); 5' GTTTTTTCTATTGCTACAAACGCGTACGCTGAGGTTCAGCTG-3' (SEQ ID NO:39); and 5'-
GCCTCCACCAAGGGCCCATCGGTCTTCCCCCTGGCACCCTCC-3' (SEQ ID NO:40), incorporating restriction sites EcoRV and Kpnl for the light chain and BsiWl and Apal for the heavy chain. Heavy and light chains were ligated into a pre-digested phage display vector (phagemid) and expressed as a monovalent Fab fragment with the heavy chain fused to the Ml 3 gene-3 minor coat protein (p3). In order to minimize deleterious effects of mutations on expression in Fab-phage format and affinity for E2 protein, single CDR libraries were constructed. To avoid contamination of the final library with wild-type sequences, templates for library construction were produced containing a TAA stop codon in each CDR targeted for mutagenesis. These templates were made by oligonucleotide-directed mutagenesis using oligonucleotides: 5'- GAAAGTGTTGATGGTTAAGGCAATAGTTTTCTG-3' (SEQ ID NO:41); 5 ' -CTGATCTATCTTGCATAAAACCTAAACTC-3 ' (SEQ ID NO:42); 5'- CAGCAAAATAATTAATAAGTGGACCCGTG-3' (SEQ ID NO:43); 5'- CCATCACTAGTGGTTAATGGAACTGGATC-3' (SEQ ID NO:44); 5'- GGATACATAAGTTAAAGTGGTAGCACTTAC-3' (SEQ ID NO:45); 5'- CTACGACTACCTAAGCTATGGACTACTGG-3 ' (SEQ ID NO:46). Six separate single CDR sub-libraries were made using oligonucleotides: 5'-
CACTTGCARAKCCKCCGAMKCTRTTGAMGSTTWTGSCRATKCTTWTMTGMA CTGG-3'(SEQ ID NO:47); 5 ' -GATCTATMTTKCAKCCRACMTARACKCTGGG-3 ' (SEQ ID NO:48); 5'- CTACTGTS AGS AARATRATRTTGAMSCCTKGASTTTTGG-3 ' (SEQ ID NO:49); 5'- CTCTGSTGAMKCCRTCASTKCTGSTTWCTKGRACTGG-3'(SEQ ID NO:50); 5'- GGGATWCRTAKCTTWCKCTGSTKCCTWCTWCRATMTAKCTMT CARAKCTCGG-3'(SEQ ID NO:51); and 5'-
CGCTGATTASTASGASTASCTWTKCTMTGGAMTWCTGG-3 ' (SEQ ID NO:52). Letter codes were used to allow multiple codons at designated positions using the key in Table 1. Solution-phase sorting strategies, with increasing stringency via decreasing concentration of
biotinylated target, similar to that described in Lee et al. J Mol Biol. (2004), 340: 1073-93, were employed except that semi-automated panning of the libraries was done on a Kingfisher instrument (Thermo Scientific). sE266i was recombinantly expressed in SF9 insect cells, purified and biotinylated at reactive ε-amino groups of lysine residues using Sulfo-NHS-SS- Biotin (Pierce) according to the manufacturer's instructions. Incubations of a phage library with the target were performed in a buffer containing 3% milk PBS/0.01 Polysorbate-20, washed with the same buffer and phage was eluted from beads using 0.1M DTT. Phage eluted from each round of selection were propagated for the next round by growth in XL-1 Blue cells (Agilent Technologies), Selection rounds alternated between sE266i produced from HCV genotype la or genotype lb sequences. Selected clones were ranked by competitive phage-binding as previously described in Lee et al. J Mol Biol. (2004), 340: 1073- 93.
Table 1. Mixed Sites IUB DNA Codes

[0263] Anti-HCV antibody variants of humanized MRCT10.1.1 were prepared by altering specific heavy and light chain CDR residues based on affinity changes selected by phage display. VL-N30Y and VH-T98S were residue alterations shown to improve affinity and were used to generate V317, V361, and V362 (Figure 2 and Table 2). In two variants (VI and V362) a potential N-glycosylation site in heavy chain CDR-H2 was removed by mutating Asn60 to serine. Also, as Asn-Leu-Ser represented a motif where N-glycosylation could occur, variants were made with SLS (e.g., VI and V362) and NPS (e.g., V6, V335, and V361) at wild type positions N60, L61, S62 according to Kabat numbering of heavy chain CDR-H2 (Table 2). Variants were also made by altering sites of the light chain with potential molecular instability. Specifically, an Asp-Gly motif (D27c and G27d according to Kabat numbering) with possible isomerization capability was removed by mutating CDR-L1 D27c
to serine (S). Additional mutations were made to improve binding affinity to soluble E2 protein (Figure 2, Tables 2, 3, and 4).
Table 2. Amino acid changes in anti-HCV antibody variants as compared to MRCTl 0.1.1

Improved binding affinity
Removed potential glycosylation site
Removed potential isomerization site
Table 3. Heavy chain hypervariable regions (HVRs)

GDSrrSGYWN YISYSGSTYYNLSLRS ALITTSTYAMDY
V355
(SEQ ID NO: 7) (SEQ ID NO: 11) (SEQ ID NO: 12)
GDSrrSGYWN YISYSGSTYYNLSLRS ALITTTTYAMDY
MRCTlO.l
(SEQ ID NO: 7) (SEQ ID NO: 11) (SEQ ID NO: 13)
Table 4. Light chain hypervariable regions (HVRs)

[0264] Oligonucleotide-directed mutagenesis was used to introduce amino acid
substitutions in the CDRs of MRCTlO. l using single- stranded DNA (ssDNA) prepared in E. coli strain CJ236 (dut-ung-) as a template. See Kunkel et al., PNAS, (1985), 82(2):488-492. For preparation of MRCTlO. l single stranded DNA, transformation-competent CJ236 cells were transformed with MRCTlO. l heavy or light chain DNA using the protocol of Chung et
al., Nucleic Acids Res., (1988), 16: 3580. A single colony was picked from growth of the transformation mixture on LB-agar plates containing 50 μg/mL carbeniciUin and was used to inoculate 1 mL of 2YT broth containing 50 μg/mL carbeniciUin. The culture was grown at 37 °C for 6 to 8 hours with continuous shaking, A 10 μΐ. aliquot of 1012 pfu/mL M13K07 helper phage (New England Biolabs) was added to the culture and incubation with shaking was continued at 37°C for 15 min before 1 mL of the culture was transferred to 50 mL of 2YT broth containing 50 μg/mL carbeniciUin and 50 μg/mL kanamycin. The culture was grown overnight at 37°C and the culture was subsequently centrifuged at 6,000 rpm for 10 minutes to pellet cells from the supernatant. The supernatant was collected and subjected to two cycles of polyethylene glycol (PEG) precipitation. Each PEG precipitation cycle consisted of resuspending the supernatant in a 1/5 volume of 20% PEG/2.5 M NaCl solution to precipitate phage particles followed by incubation for 10 minutes at room temperature, centrifugation at 13,000 rpm for 15 minutes to collect the phage pellet, re-suspension of the phage pellet in phosphate buffered saline (PBS) before centrifugation at 13,000 rpm for 15 minutes. After PEG precipitation, the phage pellet was re-suspended in 1 mL PBS for preparation of ssDNA from the phage pellet using a Qiagen Ml 3 spin kit according to the manufacturer's instructions. After elution of the ssDNA from the spin column with 100 μΐ^ elution buffer, the absorbance at 260 nm was measured to determine MRCT10.1 ssDNA concentration where an A260 reading of 1 was equivalent to 33 rιg/μL· ssDNA. For amino acid mutagenesis, specific oligonucleotides (330 rιg/μL· stock concentration) for construction of each variant were synthesized using standard chemical methods (Table 5). The oligonucleotides were phosphorylated by preparing a solution containing 2 μL·
oligonucleotide, 2 μΐ, ΙΟχ TM (0.5 M Tris-HCl pH 7.5, 0.1 M MgCl2) buffer, 2 μΐ, 10 mM rATP, 1 μΐ, 100 mM DTT, and 12 μΐ, H20. A 1 μΐ, aliquot of T4 polynucleotide kinase (New England BioLabs) was added and the solution was incubated at 37°C for 30 min. A 2 μL· aliquot of phosphorylated oligonucleotide was annealed with 1 μg MRCT10.1 ssDNA in a solution containing 2.5 μΐ^ ΙΟχ TM buffer and an amount of H20 that brought the total volume to 25 μΐ^. The mixture was incubated for 1 minute at 90°C then 5 minutes at 50°C before being placed on ice. The reaction was initiated by adding 1 μί 10 mM rATP, 1 μL· 25 mM dNTP mix (equal volumes of 100 mM each dATP, dCTP, dTTP, dGTP), 1.5 μΐ, 100 mM DTT, 0.6 μΐ, T4 DNA ligase (New England BioLabs), 0.3 μΐ, T7 DNA Polymerase (New England BioLabs) and the mixture was incubated at 37°C for 1.5 hours. A 1 μL· portion of the mutagenesis reaction was used to transform XL- 1 Blue (Stratagene) using the manufacturer's protocol and the transformants were plated on LB agar plates containing 50
μg/mL carbeniciUin. After overnight growth at 37 °C, 4 to 6 single colonies were picked and grown overnight in 5 mLs of LB broth containing 50 μg/mL carbeneciUin. After incubation, dsDNA was prepared using a Qiagen mini-prep spin kit (Qiagen) and the correct sequence clones were identified using dideoxynucleotide sequencing.
Table 5. Oligonucleotides for site-directed mutagenesis

Variable regions were amplified by PCR using oligonucleotides: forward primer 5'- CTCGGTTCTATCG ATTG A ATTCC ACC ATGGG- 3 ' (SEQ ID NO:59) and reverse primer 5 ' -GCC AAGGGACCACGGTC ACCGTCTCCTC AG-3 ' (SEQ ID NO:60) for heavy chain and forward primer 5'- GGAGTACATTCAGATATCGTGTTGACGC-3 ' (SEQ ID NO:61) and reverse primer 5'- CTGGAGATCAAACGGACCGTGGCTGCAC -3' (SEQ ID NO:62) for light chain to incorporate restriction sites for directional cloning. Light chain variable domains (VL) were digested with EcoRY and rsrll. Heavy chain variable domains (VH) were digested with Clal and BstEII. Digested variable region fragments were ligated into pre-digested pRK5 vectors encoding human kappa I antibody for the light chain, or human IgGl antibody for the heavy chain, and the ligation product was transformed into Escherichia coli strain XL 1 -blue (Strategene, La Jolla, CA, USA). Single colonies were selected for plasmid purification. Correctly subcloned plasmids were determined by DNA sequencing.
[0266] Co-transfection of heavy and light chain plasmids into 293T or CHO cells was used for recombinant expression of the antibody variants. For variant antibody expression from 293T cells, 0.1 mL FuGENE 6 transfection reagent (Roche Applied Science, Mannheim, Germany) and 5 μg each of the heavy and light chain vectors (10 μg total endo toxin-free DNA) in a final volume of 1 mL transfection media (Freestyle™ 293 Expression Medium) was prepared for transfection. FuGENE™ 6-DNA complexes were allowed to form for a minimum of 20 minutes before adding to a T150 flask containing 293T cells in a final volume of 25 mL transfection media. A 1 mL aliquot of transfection mixture was added to each T150 flask and the cells were incubated. After incubation, conditioned media was collected 4 to 7 days post-transfection. PMSF and bovine lung aprotinin were added to final concentrations of 1 mM and 1.2 μg/mL, respectively. The media was filtered using a low protein binding polystyrene bottle with a 0.22 μιη cellulose acetate filter to remove detached cells. IgG was purified on a 0.5 to 1 mL of rProtein A agarose column. The column was washed with 25 mL of 75 mM Tris, 1.5M KC1, pH 8 or PBS, pH7.4, before eluting with 2.5 mL 100 mM acetic acid, 150 mM NaCl into fractions in tubes containing a 1/10 volume of 1M Tris, pH 8 to neutralize the solution. Purified IgG was buffer exchanged into PBS using a PD-10 desalting column, followed by concentration using a Centricon-10.
Example 2: Binding affinities of anti-HCV antibodies
[0267] The generated anti-HCV antibodies were compared to wild- type MRCT10.1 for affinity. For determination of binding affinity to HCV E2 protein (sE2661), surface plasmon resonance (SPR) measurements on a Biacore A 100 instrument (GE Healthcare Biosciences AB) was used to measure kinetics of antibody variants binding to soluble E2 extracellular domain. Using a human antibody capture kit (GE Healthcare Biosciences AB), a standard coupling protocol was used to immobilize human Fc-specific IgG via primary amine coupling at 25°C. Using HBS-EP running buffer (lOmM Hepes pH 7.4, 150mM NaCl, 3mM EDTA, 0.01 polysorbate 20), IgG variants were captured in each automated cycle with a contact time of 3 minutes at a flow rate of ΙΟμΙ/min. Soluble E2 antigen was injected for 2 minutes at a flow rate of 20μ1/ηιίη in a concentration series ranging from 200nM to 0.78nM in 2-fold increments. Four flow cells were utilized simultaneously to capture antibody on spots 1 and 5, with spots 2 and 4 reserved as internal references containing only the capturing IgG.
Surfaces were regenerated after each cycle of capture and antigen binding by a 30 second injection of 3M MgCl2 solution as described in the capture kit product information brochure (GE Healthcare Biosciences AB, Human Antibody Capture Kit BR- 1008-39). Signal recorded for injection of buffer alone (zero soluble E2 concentration) was subtracted from the observed binding curves. Sensorgrams were analyzed according to a 1: 1 binding Langmuir binding model using software supplied by the manufacturer in order to calculate kinetic constants for binding of soluble E2 to Fc-captured antibody (Table 6). Further analysis of V362 demonstrated that affinity improvement resulted primarily from a decreased off-rate (Table 7)
Table 6. Affinity (KD) of anti-HCV antibodies for soluble E2 protein from HCV genotype la (GTla) and lb (GTlb).

V361 4.9 6.2
V362 2.1 2.1
N/A=Not available
Table 7. Affinity of V362 against sE266i
GTla GTlb
Antibodies Kon K0ff KD Kon K0ff KD
(xl05 MV) (xl0~3 s"1) (nM) (xl05 MV) (xl0~3 s"1) (nM)
MRCT10. 4.1 + 1.8 2.4 + 0.1 6.6 + 3.2 4.2 4.2 4.2
1
V362 3.3 + 0.5 0.9 + 0.1 2.7 + 0.5 3.6 + 0.4 0.9 + 0.1 2.6 + 0.5
Example 3: Neutralization of HCV by anti-HCV antibodies
[0268] Neutralization of HCV pseudoparticles (HCVpp) by anti-HCV antibodies VI, V6, V79, V317, V335, V355, V361, and V362 was analyzed using a neutralization assay with HCVpp stocks as previously described in Hotzel et al., Protein Eng Des Sel. (2011), 24:679- 89 and Diao et al., J Virol. (2012), 86(20): 10935-49, which are incorporated herein by reference in their entirety. HCVpp stocks were generated as previously described in Kapadia et al. J Virol, (2007), 81:374-383. Briefly, HEK 293T cells were co-transfected with the Delta 8.9 plasmid containing gag-pol, the CMV-Luc-DsRed lentiviral transfer construct, and a plasmid expressing HCV E1E2 sequences from genotype 2a (J6CF) using Lipofectamine 2000, according to the manufacturer's instructions. Forty-eight hours following transfection, the medium containing HCVpp was collected, clarified, filtered through 0.45-μιη pore-sized membranes and used for infection of Huh-7.5 cells. HCVpp stocks were normalized for luciferase activity before infecting cells. Neutralization assays using HCVpp viruses were performed in 96-well plates (Costar). Briefly, 5x10 Huh-7.5 cells were seeded in a 96-well plate 16 hours prior to the neutralization assay. The following day, three-fold dilutions of antibody were incubated with naive Huh-7.5 cells for 1 hour at 37°C, followed by the addition of HCVpp for an additional 4 hours. Unbound virus was removed by replacing with fresh DMEM and the cells were maintained in culture for 3 days before measuring HCV infection. HCVpp infection was determined using the Luciferase Assay System (Promega), according to the manufacturer's instructions.
[0269] As compared to MRCT10.1, antibody variants had significantly improved in vitro neutralization potencies against HCVpp generated from HCV genotypes la (GTla), lb (GTlb) and 2a (GT2a) (Table 8).
Table 8. HCVpp neutralization by anti-HCV antibodies
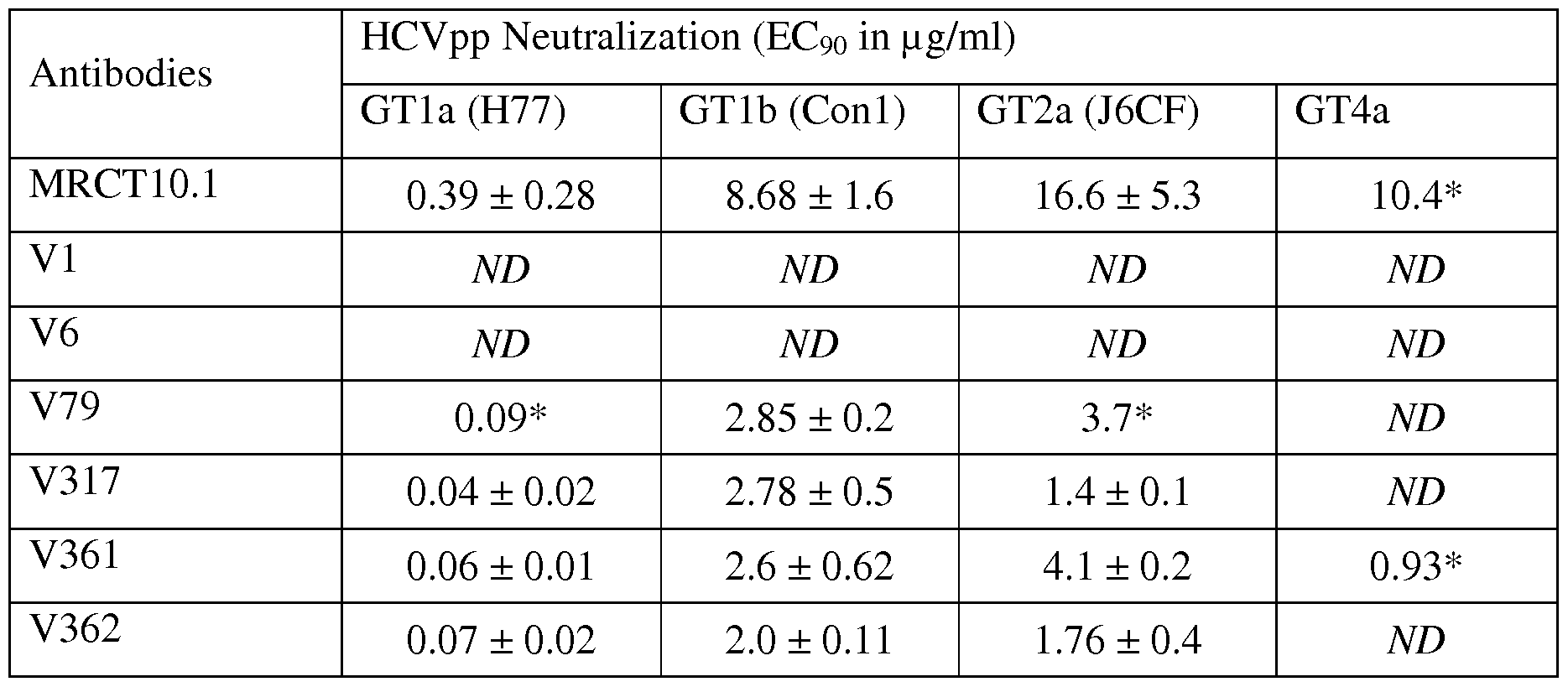
Values represent average EC90 values + SEM and are representative of at least two
independent experiments each done in triplicate. Asterisks denote neutralization data representative of a single experiment. ND = not done.
[0270] Anti-HCV antibodies were also assayed for neutralization of infectious cell culture HCV (HCVcc) of wild- type genotype 2a Jcl in addition to E2 mutants of genotype 2a Jcl that arise when treated with other anti-HCV antibodies that target the same epitope of the E2 protein. For the HCVcc neutralization assay, Jcl HCVcc virus stocks were generated by transfecting Huh7.5 cells with in vitro transcribed full-length HCV RNA as previously described in Zhong et al., PNAS, (2005), 102:9294-9, which is incorporated herein by reference in its entirety. Briefly, genotype 2a Jcl genome were chemically synthesized using sequences genotype 2a JFH-1 (GenBank accession number AB047639) and genotype 2a J6CF (GenBank accession number AF177036) based on the chimeric strategy as described in Pietschmann et al., PNAS, (2006), 103:7408-7413 and cloned into pUC19 plasmid (pUC Jcl). Plasmids were linearized with Xbal and in vitro transcribed RNA was generated using Megascript kit (Ambion) according to the manufacturer's instructions. HCV RNA was transfected into Huh-7.5 cells by electroporation and supernatants from Jcl RNA-transfected cells were harvested starting from 3 days post transfection, titered and pooled to generate an HCVcc stock. HCVcc neutralization assays were performed as described for the HCVpp
neutralization assay above. Huh-7.5 cells were infected with an MOI=0.3 (Multiplicity of infection (MOI) for Jcl HCVcc was calculated based on virus titer as measured by TCID50 calculations. See Lindenbach et al. Science, (2005), 309:623-626). After three days, HCVcc infection was determined by real time quantitative PCR as described in Hotzel et al., Protein Eng Des Sel. (2011), 24:679-89 and Diao et al., J Virol. (2012), 86(20): 10935-49. As compared to AP33, the murine antibody of MRCT10.1, antibody variants had significantly improved in vitro neutralization potencies against wild- type genotype 2a Jcl HCVcc (Table 9). N417S, N417T, and N415D HCVcc were completely resistant to inhibition by V362 and in the case of N417S HCVcc by V361. In contrast, the N417G HCVcc was sensitive to V362 (Table 9).
Table 9. Jcl HCVcc sensitivity to HCV antibodies

All values represent average EC90 values + SEM and are representative of at least two independent experiments each done in triplicate. Asterisks denote values determined from three independent experiments. ND = not done. WT = wild-type genotype 2a Jcl
[0271] Binding of V362 to either E241 4 J peptide or full-length E1E2 expressed in transfected cells was assayed. Binding of Fab fragments to chemically synthesized peptides (non-glycosylated) was measured by Surface plasmon resonance (SPR) or Bio-Layer
Inferometry (BLI). V362 showed measurable binding affinity to the E2 412-423 peptide epitope with a Ku of 30 nM. Ratios of binding alanine mutant versus wild-type versions of the peptide were measured to further characterize binding of V362 to the E2 epitope (Figure 3 A). V362 did not bind the W420A mutant peptide but did bind wild-type and N417A mutant peptides (Figure 3A). Mutants L413A and G418A affected V362 binding by ~8-fold and ~2- fold, respectively, and binding of V362 to N415A only decreased by 2.8-fold (Figure 3A).
Since the region surrounding the E2 412-"423 epitope is highly glycosylated on virion-associated E2, the contribution of possible post-translational modifications to epitope binding by V362 was assayed. In order to determine if V362 bound cellular expressed E2 containing the
N417S or N417T mutations, antibody binding to a soluble E2 protein variant (sE266i) or E1E2 expressed in cells using SPR and ELISA, respectively was tested. SPR measurements indicated affinity of V362 for N417S and N417T sE266i was reduced at least 100-fold (¾>500 nM) whereas binding of N417G sE266i was decreased only 3-fold. Furthermore, V362 binding to ElE2-expressing 293T cell lysates demonstrated no detectable binding to N417S, N417T or N415D with any of the E2412-423 -specific antibodies (Figure 3B). In comparison, MRCT10.1 Fab still bound the N417S peptide, although with ~3.9-fold less affinity compared to the wild- type peptide (Figure 3C and D).
Example 4: Combination of anti-HCV antibodies with HCV antivirals enhanced suppression of infection and spread of resistant HCV.
[0272] The N417S resistant mutant genotype 2a HCVcc rapidly emerges in vitro in the presence of previously described AP33 antibody. The ability of anti-HCV antibodies when used in combination with an HCV direct acting antiviral (DAA) to enhance antiviral efficacy and suppress the emergence of resistant HCVcc was assessed. To determine if V362 antibody could suppress the emergence of resistance to an approved HCV NS3 protease inhibitor (Telaprevir), DMSO-differentiated Huh-7.5 cells were infected with Jcl HCVcc alone or in the presence of Telaprevir, V362 antibody or a combination of both Telaprevir and V362 antibody (Figure 4). For the assay, Huh7.5 cells (90% confluent) were cultured in collagen coated 96-well plates in the presence of DMEM containing 1% DMSO. Culture media was replenished every two days for 14 days before cells were infected with Jcl HCVcc (MOI=0.05). One day post-infection, cells were left untreated or treated with 2 μΜ
Telaprevir or a combination of 2 μΜ Telaprevir and 10 μg/ml V362 antibody. A
concentration of 2 μΜ Telaprevir was used, which was similar to the trough concentrations detected in some treated patients. See Reesinik et al. Gastroenterology. (2006). 131:997- 1002. Fresh inhibitors were added every two days until day 22 post infection. HCV RNA replication was measured at various times post infection by RT-qPCR. Jcl HCVcc infection of DMSO-differentiated cells in the presence of Telaprevir induced early viral suppression but resulted in the emergence of a T54A HCVcc variant approximately 18 days post infection (Figure 4, open triangles). The T54A mutation has been demonstrated to confer resistance to Telaprevir and can be detected in some patients treated with Telaprevir monotherapy.
Treatment of cells with 10 μg/mL V362 antibody initially inhibited HCV infection but resulted in no significant antiviral effect 10 days post infection, consistent with the rapid
emergence of V362-resistant variants (Figure 4, open circles). In comparison, combination treatment with V362 antibody with Telaprevir resulted in undetectable HCV RNA replication even at 22 days post infection when the T54A mutant emerged as the dominant resistant species in the Telaprevir-treated cultures (Figure 4, open diamonds).
[0273] To determine if V335 antibody could suppress the emergence of resistance upon treatment with Telaprevir and interferon- alpha (IFN-a), an approved antiviral cytokine, DMSO-differentiated Huh-7.5 cells were infected with Jcl HCVcc alone or in the presence of Telaprevir and IFN-a or a combination of Telaprevir, IFN-a and V335 antibody (Figure 5A). For the assay, Huh7.5 cells (90% confluent) were cultured in collagen coated 96-well plates in the presence of DMEM containing 1% DMSO. Culture media was replenished every two days for 14 days before cells were infected with Jcl HCVcc (MOI=0.05). One day postinfection, cells were left untreated or treated with 0.3 μΜ Telaprevir plus 5 IU/ml IFN-a or a combination of 0.3 μΜ Telaprevir and 5 IU/ml IFN-a plus 10 μg/ml V335 antibody or 10 μg/ml MRCT10.1 antibody. Fresh inhibitors were added every two days until day 22 post infection. HCV RNA replication was measured at various times post infection by RT-qPCR. While treatment of Jcl HCVcc infection with Telaprevir and IFN-a resulted in significant suppression of Jcl HCV infection, viral replication rebounded to untreated levels between 2 to 3 weeks post infection (Figure 5 A, open circles). In contrast, addition of 10 μg/mL V335 antibody to the combination of Telaprevir and IFN-a resulted in significant suppression of viral replication even up to 30 days post infection (Figure 5 A, inverted filled diamonds). In addition, V335 antibody was more effective at suppressing viral infection and spread in vitro as compared to MRCT10.1 antibody (Figure 5B, inverted filled diamonds).
Claims
1. An isolated antibody that binds hepatitis C virus E2 protein comprising a heavy chain variable region and a light chain variable region,
(a) wherein the heavy chain variable region comprises three hypervariable region (HVR) sequences:
(i) HVR-H1 comprising GXiSX2TSGYWN (SEQ ID NO: 1), wherein Xi is D or E, and X2 is I or L;
(ii) HVR-H2 comprising YISYSGSTYYXiX2SLRS (SEQ ID NO:2), wherein Χ is N or S, and X2 is P or L; and
(iii) HVR-H3 comprising ALITTX \ T Y AMD Y (SEQ ID NO:3), wherein Xi is S or T; and
(b) wherein the light chain variable region comprises three HVR sequences:
(iv) HVR-L1 comprising RASES VX1GYGX2SFLH (SEQ ID NO:4), wherein Xjis D or S, and X2= N or Y;
(v) HVR-L2 comprising LASNLNS (SEQ ID NO:5); and
(vi) HVR-L3 comprising QQNNVDPWT (SEQ ID NO:6);
wherein the antibody is not an antibody comprising (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:7; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 11 ; (c) HVR-H3 comprising the amino acid sequence of SEQ ID NO: 13; (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 15; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:5; and (f) HVR-L3 comprising an amino acid sequence selected from SEQ ID NO:6.
2. The antibody of claim 1, wherein the antibody comprises HVR-H1 comprising the amino acid sequence of SEQ ID NO:7 or 8, HVR-H2 comprising the amino acid sequence of SEQ ID NO:9, 10 or 1 1, and HVR-H3 comprising the amino acid sequence of SEQ ID NO: 12 or 13.
3. The antibody of claim 1 or 2, wherein the antibody comprises HVR-L1 comprising the amino acid sequence of SEQ ID NO: 14, 15 or 16, HVR-L2 comprising the amino acid sequence of SEQ ID NO:5, and HVR-L3 comprising the amino acid sequence of SEQ ID NO:6.
4. The antibody of claim 1, wherein the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:7, HVR-H2 comprising the amino acid
sequence of SEQ ID NO:9 or 10, and HVR-H3 comprising the amino acid sequence of SEQ ID NO: 12; and/or (b) HVR-Ll comprising the amino acid sequence of SEQ ID NO: 14, HVR- L2 comprising the amino acid sequence of SEQ ID NO:5, and HVR-L3 comprising the amino acid sequence of SEQ ID NO: 6.
5. The antibody of claim 1, wherein the antibody comprises the heavy chain variable region sequence of SEQ ID NO: 17, 18, or 22, and/or the light chain variable region sequence of SEQ ID NO:24.
6. The antibody of claim 1, wherein the antibody comprises the heavy chain sequence of SEQ ID NO:27 or 28, and the light chain sequence of SEQ ID NO:30.
7. An isolated antibody that binds hepatitis C virus E2 protein comprising a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO:9 or 10, CDR H3 comprising the amino acid sequence of SEQ ID NO:34; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
8. The antibody of any one of claims 1-7, wherein the antibody is a humanized antibody.
9. The antibody of any one of claims 1-7, wherein the antibody is an antigen- binding fragment.
10. The antibody of any one of claims 1-7, wherein the antibody is an antibody fragment selected from the group consisting of a Fab, Fab'-SH, Fv, scFv, and (Fab')2 fragment.
11. The antibody of any one of claims 1-10, wherein the antibody inhibits HCV genotype la, genotype lb, genotype 2a, and genotype 4a infection to liver cells.
12. An antibody that binds hepatitis C virus E2 protein produced by a method comprising culturing a host cell comprising a nucleic acid encoding the antibody of any one of claims 1-11 under a condition suitable for expression of the nucleic acid; and recovering the antibody produced by the cell.
13. A pharmaceutical composition comprising the antibody of any one of claims 1-12, and a pharmaceutically acceptable carrier.
14. An isolated nucleic acid encoding the antibody of any one of claims 1-11.
15. A vector comprising a nucleic acid encoding the antibody of any one of claims
1-11.
16. The vector of claim 15, wherein the vector is an expression vector.
17. A host cell comprising a nucleic acid encoding the antibody of any one of claims 1-11.
18. The host cell of claim 17, wherein the host cell is prokaryotic or eukaryotic.
19. A method of producing an antibody comprising culturing a host cell comprising a nucleic acid encoding the antibody of any one of claims 1-11 under a condition suitable for expression of the nucleic acid; and recovering the antibody produced by the cell.
20. The method of claim 19, further comprising purifying the antibody.
21. A method for treating or preventing a hepatitis C virus infection in a subject, comprising administering to the subject an effective amount of the antibody of any one of claims 1-12.
22. The method of claim 21, wherein the subject is a human.
23. The method of claim 21, wherein the subject has been diagnosed with the hepatitis C virus infection.
24. The method of claim 21, wherein the hepatitis C virus infection is an acute hepatitis C virus infection.
25. The method of claim 21, wherein the hepatitis C virus infection is a chronic hepatitis C virus infection.
26. The method of claim 21, wherein treating the hepatitis C virus infection comprises reducing viral load.
27. The method of claim 21, further comprising administering a second therapeutic agent.
28. The method of claim 27, wherein the second therapeutic agent is a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a cyclophilin inhibitor, an antibody that targets a non-E2 HCV protein, or a combination thereof.
29. The method of claim 27, wherein the second therapeutic agent is a HCV protease inhibitor selected from the group consisting of Teleprevir, Boceprevir, Semeprevir (TMC435), BI201335, Danoprevir/r (RG7227), Vaniprevir (MK-7009), Narlaprevir (SCH 900518), Asunaprevir, GS-9256, GS-9451, ABT-450/r, ACH-1625, ACH-2684, and MK- 5172.
30. The method of claim 27, wherein the second therapeutic agent is a polymerase inhibitor selected from the group consisting of PSI-7977, Mercitabine, IDX184, PSI-938, INX- 189, Tegobuvir, Filibuvir, Setrobuvir, BI207127, ABT-333, VX-222, and TMC-647055.
31. The method of any one of claims 21 - 30, wherein the treatment is an interferon-free treatment.
32. The method of any one of claims 21 - 30, wherein the subject has sustained virologic response for at least 12 weeks after stopping the treatment.
33. The method of any one of claims 21 - 30, wherein the viral load in the subject has been reduced to an undetectable level after the treatment.
34. The method of any one of claims 21 - 30, wherein the viral resistance in the subject is undetectable or low.
35. The method of any one of claims 21 - 30, wherein the subject is not responsive to an interferon treatment.
36. A method of preventing developing resistance to treatment, comprising administering an effective amount of an antibody that specifically binds hepatitis C virus E2 protein and a second therapeutic agent, wherein the second therapeutic agent is a viral protease inhibitor, a viral polymerase inhibitor, an NS5A inhibitor, an interferon, a cyclophilin inhibitor, an antibody that targets a non-E2 HCV protein, and a combination thereof.
37. The method of claim 36, wherein the antibody comprises a heavy chain variable region and a light chain variable region,
(a) wherein the heavy chain variable region comprises three hypervariable region (HVR) sequences:
(i) HVR-H1 comprising GXiSX2TSGYWN (SEQ ID NO: 1), wherein Χ is D or E, and X2 is I or L;
(ii) HVR-H2 comprising YISYSGSTYYXiX2SLRS (SEQ ID NO:2), wherein Xi is N or S, and X2 is P or L; and
(iii) HVR-H3 comprising ALITTX \ T Y AMD Y (SEQ ID NO:3), wherein Χ is S or T; and
(b) wherein the light chain variable region comprises three HVR sequences:
(iv) HVR-L1 comprising RASES VXiGYGX2SFLH (SEQ ID NO:4), wherein Xiis D or S, and X2= N or Y;
(v) HVR-L2 comprising LASNLNS (SEQ ID NO:5); and
(vi) HVR-L3 comprising QQNNVDPWT (SEQ ID NO:6).
38. The method of claim 36, wherein the antibody comprising a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises CDR HI comprising the amino acid sequence of SEQ ID NO:33, CDR H2 comprising the amino acid sequence of SEQ ID NO:9, 10, or 11, CDR H3 comprising the amino acid sequence of SEQ ID NO:34 or 35; and wherein the light chain variable region comprises CDR LI comprising the amino acid sequence of SEQ ID NO: 14, 15, or 16, CDR L2 comprising the amino acid sequence of SEQ ID NO:5, and CDR L3 comprising the amino acid sequence of SEQ ID NO:6.
39. A method of preventing of HCV infection of a transplanted liver, comprising administering to the subject an effective amount of the antibody of any one of claims 1-12 before, during or after the subject receives the liver transplant.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361755932P | 2013-01-23 | 2013-01-23 | |
| US61/755,932 | 2013-01-23 | ||
| US201361770272P | 2013-02-27 | 2013-02-27 | |
| US61/770,272 | 2013-02-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014116749A1 true WO2014116749A1 (en) | 2014-07-31 |
Family
ID=50073501
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2014/012610 WO2014116749A1 (en) | 2013-01-23 | 2014-01-22 | Anti-hcv antibodies and methods of using thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2014116749A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022153898A1 (en) * | 2021-01-13 | 2022-07-21 | デンカ株式会社 | Method for measuring target antigen, and insoluble particles and target antigen measurement kit used therein |
Citations (100)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3773919A (en) | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| USRE30985E (en) | 1978-01-01 | 1982-06-29 | Serum-free cell culture media | |
| EP0073657A1 (en) | 1981-08-31 | 1983-03-09 | Genentech, Inc. | Preparation of hepatitis B surface antigen in yeast |
| US4419446A (en) | 1980-12-31 | 1983-12-06 | The United States Of America As Represented By The Department Of Health And Human Services | Recombinant DNA process utilizing a papilloma virus DNA as a vector |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| US4601978A (en) | 1982-11-24 | 1986-07-22 | The Regents Of The University Of California | Mammalian metallothionein promoter system |
| WO1987000195A1 (en) | 1985-06-28 | 1987-01-15 | Celltech Limited | Animal cell culture |
| US4657866A (en) | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US4737456A (en) | 1985-05-09 | 1988-04-12 | Syntex (U.S.A.) Inc. | Reducing interference in ligand-receptor binding assays |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| WO1990003430A1 (en) | 1988-09-23 | 1990-04-05 | Cetus Corporation | Cell culture medium for enhanced cell growth, culture longevity and product expression |
| US4927762A (en) | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| US4965199A (en) | 1984-04-20 | 1990-10-23 | Genentech, Inc. | Preparation of functional human factor VIII in mammalian cells using methotrexate based selection |
| WO1990013646A1 (en) | 1989-04-28 | 1990-11-15 | Transgene S.A. | Application of novel dna fragments as a coding sequence for a signal peptide for the secretion of mature proteins by recombinant yeast, expression cassettes, transformed yeasts and corresponding process for the preparation of proteins |
| EP0404097A2 (en) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispecific and oligospecific, mono- and oligovalent receptors, production and applications thereof |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| WO1993001161A1 (en) | 1991-07-11 | 1993-01-21 | Pfizer Limited | Process for preparing sertraline intermediates |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| WO1993016185A2 (en) | 1992-02-06 | 1993-08-19 | Creative Biomolecules, Inc. | Biosynthetic binding protein for cancer marker |
| WO1994011026A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Therapeutic application of chimeric and radiolabeled antibodies to human b lymphocyte restricted differentiation antigen for treatment of b cell lymphoma |
| WO1994029351A2 (en) | 1993-06-16 | 1994-12-22 | Celltech Limited | Antibodies |
| US5500362A (en) | 1987-01-08 | 1996-03-19 | Xoma Corporation | Chimeric antibody with specificity to human B cell surface antigen |
| US5571894A (en) | 1991-02-05 | 1996-11-05 | Ciba-Geigy Corporation | Recombinant antibodies specific for a growth factor receptor |
| US5587458A (en) | 1991-10-07 | 1996-12-24 | Aronex Pharmaceuticals, Inc. | Anti-erbB-2 antibodies, combinations thereof, and therapeutic and diagnostic uses thereof |
| US5624821A (en) | 1987-03-18 | 1997-04-29 | Scotgen Biopharmaceuticals Incorporated | Antibodies with altered effector functions |
| US5648237A (en) | 1991-09-19 | 1997-07-15 | Genentech, Inc. | Expression of functional antibody fragments |
| WO1997030087A1 (en) | 1996-02-16 | 1997-08-21 | Glaxo Group Limited | Preparation of glycosylated antibodies |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US5750373A (en) | 1990-12-03 | 1998-05-12 | Genentech, Inc. | Enrichment method for variant proteins having altered binding properties, M13 phagemids, and growth hormone variants |
| US5770429A (en) | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5789199A (en) | 1994-11-03 | 1998-08-04 | Genentech, Inc. | Process for bacterial production of polypeptides |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| US5840523A (en) | 1995-03-01 | 1998-11-24 | Genetech, Inc. | Methods and compositions for secretion of heterologous polypeptides |
| WO1998058964A1 (en) | 1997-06-24 | 1998-12-30 | Genentech, Inc. | Methods and compositions for galactosylated glycoproteins |
| US5869046A (en) | 1995-04-14 | 1999-02-09 | Genentech, Inc. | Altered polypeptides with increased half-life |
| WO1999022764A1 (en) | 1997-10-31 | 1999-05-14 | Genentech, Inc. | Methods and compositions comprising glycoprotein glycoforms |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| WO1999051642A1 (en) | 1998-04-02 | 1999-10-14 | Genentech, Inc. | Antibody variants and fragments thereof |
| US6040498A (en) | 1998-08-11 | 2000-03-21 | North Caroline State University | Genetically engineered duckweed |
| US6075181A (en) | 1990-01-12 | 2000-06-13 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO2000061739A1 (en) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US6171586B1 (en) | 1997-06-13 | 2001-01-09 | Genentech, Inc. | Antibody formulation |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| WO2001029246A1 (en) | 1999-10-19 | 2001-04-26 | Kyowa Hakko Kogyo Co., Ltd. | Process for producing polypeptide |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| US6267958B1 (en) | 1995-07-27 | 2001-07-31 | Genentech, Inc. | Protein formulation |
| WO2002031140A1 (en) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cells producing antibody compositions |
| US6420548B1 (en) | 1999-10-04 | 2002-07-16 | Medicago Inc. | Method for regulating transcription of foreign genes |
| US20020164328A1 (en) | 2000-10-06 | 2002-11-07 | Toyohide Shinkawa | Process for purifying antibody |
| WO2003011878A2 (en) | 2001-08-03 | 2003-02-13 | Glycart Biotechnology Ag | Antibody glycosylation variants having increased antibody-dependent cellular cytotoxicity |
| US20030115614A1 (en) | 2000-10-06 | 2003-06-19 | Yutaka Kanda | Antibody composition-producing cell |
| US6602684B1 (en) | 1998-04-20 | 2003-08-05 | Glycart Biotechnology Ag | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US20030157108A1 (en) | 2001-10-25 | 2003-08-21 | Genentech, Inc. | Glycoprotein compositions |
| WO2003085107A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Cells with modified genome |
| WO2003084570A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | DRUG CONTAINING ANTIBODY COMPOSITION APPROPRIATE FOR PATIENT SUFFERING FROM FcϜRIIIa POLYMORPHISM |
| WO2003085119A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | METHOD OF ENHANCING ACTIVITY OF ANTIBODY COMPOSITION OF BINDING TO FcϜ RECEPTOR IIIa |
| US20040093621A1 (en) | 2001-12-25 | 2004-05-13 | Kyowa Hakko Kogyo Co., Ltd | Antibody composition which specifically binds to CD20 |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US20040110282A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells in which activity of the protein involved in transportation of GDP-fucose is reduced or lost |
| US20040109865A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Antibody composition-containing medicament |
| US20040132140A1 (en) | 2002-04-09 | 2004-07-08 | Kyowa Hakko Kogyo Co., Ltd. | Production process for antibody composition |
| WO2004056312A2 (en) | 2002-12-16 | 2004-07-08 | Genentech, Inc. | Immunoglobulin variants and uses thereof |
| US20050014934A1 (en) | 2002-10-15 | 2005-01-20 | Hinton Paul R. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| US20050079574A1 (en) | 2003-01-16 | 2005-04-14 | Genentech, Inc. | Synthetic antibody phage libraries |
| WO2005035778A1 (en) | 2003-10-09 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | PROCESS FOR PRODUCING ANTIBODY COMPOSITION BY USING RNA INHIBITING THE FUNCTION OF α1,6-FUCOSYLTRANSFERASE |
| WO2005035586A1 (en) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | Fused protein composition |
| US20050119455A1 (en) | 2002-06-03 | 2005-06-02 | Genentech, Inc. | Synthetic antibody phage libraries |
| US20050123546A1 (en) | 2003-11-05 | 2005-06-09 | Glycart Biotechnology Ag | Antigen binding molecules with increased Fc receptor binding affinity and effector function |
| WO2005053742A1 (en) | 2003-12-04 | 2005-06-16 | Kyowa Hakko Kogyo Co., Ltd. | Medicine containing antibody composition |
| WO2005100402A1 (en) | 2004-04-13 | 2005-10-27 | F.Hoffmann-La Roche Ag | Anti-p-selectin antibodies |
| US20050260186A1 (en) | 2003-03-05 | 2005-11-24 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US20050266000A1 (en) | 2004-04-09 | 2005-12-01 | Genentech, Inc. | Variable domain library and uses |
| US6982321B2 (en) | 1986-03-27 | 2006-01-03 | Medical Research Council | Altered antibodies |
| US20060025576A1 (en) | 2000-04-11 | 2006-02-02 | Genentech, Inc. | Multivalent antibodies and uses therefor |
| WO2006029879A2 (en) | 2004-09-17 | 2006-03-23 | F.Hoffmann-La Roche Ag | Anti-ox40l antibodies |
| WO2006044908A2 (en) | 2004-10-20 | 2006-04-27 | Genentech, Inc. | Antibody formulation in histidine-acetate buffer |
| US7041870B2 (en) | 2000-11-30 | 2006-05-09 | Medarex, Inc. | Transgenic transchromosomal rodents for making human antibodies |
| US20060104968A1 (en) | 2003-03-05 | 2006-05-18 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminogly ycanases |
| US7087409B2 (en) | 1997-12-05 | 2006-08-08 | The Scripps Research Institute | Humanization of murine antibody |
| WO2006100449A1 (en) | 2005-03-19 | 2006-09-28 | Medical Research Council | Improvements in or relating to treatment and prevention of viral infections |
| US7125978B1 (en) | 1999-10-04 | 2006-10-24 | Medicago Inc. | Promoter for regulating expression of foreign genes |
| US7189826B2 (en) | 1997-11-24 | 2007-03-13 | Institute For Human Genetics And Biochemistry | Monoclonal human natural antibodies |
| US20070061900A1 (en) | 2000-10-31 | 2007-03-15 | Murphy Andrew J | Methods of modifying eukaryotic cells |
| US20070117126A1 (en) | 1999-12-15 | 2007-05-24 | Genentech, Inc. | Shotgun scanning |
| US20070160598A1 (en) | 2005-11-07 | 2007-07-12 | Dennis Mark S | Binding polypeptides with diversified and consensus vh/vl hypervariable sequences |
| US20070237764A1 (en) | 2005-12-02 | 2007-10-11 | Genentech, Inc. | Binding polypeptides with restricted diversity sequences |
| US20070292936A1 (en) | 2006-05-09 | 2007-12-20 | Genentech, Inc. | Binding polypeptides with optimized scaffolds |
| US20080069820A1 (en) | 2006-08-30 | 2008-03-20 | Genentech, Inc. | Multispecific antibodies |
| US7371826B2 (en) | 1999-01-15 | 2008-05-13 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2008077546A1 (en) | 2006-12-22 | 2008-07-03 | F. Hoffmann-La Roche Ag | Antibodies against insulin-like growth factor i receptor and uses thereof |
| US20090002360A1 (en) | 2007-05-25 | 2009-01-01 | Innolux Display Corp. | Liquid crystal display device and method for driving same |
| US7521541B2 (en) | 2004-09-23 | 2009-04-21 | Genetech Inc. | Cysteine engineered antibodies and conjugates |
| US7527791B2 (en) | 2004-03-31 | 2009-05-05 | Genentech, Inc. | Humanized anti-TGF-beta antibodies |
| WO2009081285A2 (en) | 2007-12-17 | 2009-07-02 | Medical Research Council Technology | Hepatitis c virus antibodies |
| WO2009089004A1 (en) | 2008-01-07 | 2009-07-16 | Amgen Inc. | Method for making antibody fc-heterodimeric molecules using electrostatic steering effects |
| WO2010080528A1 (en) * | 2008-12-17 | 2010-07-15 | Genentech, Inc. | Hepatitis c virus combination therapy |
| WO2011147863A1 (en) * | 2010-05-25 | 2011-12-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Combination of anti-envelope antibodies and anti-receptor antibodies for the treatment and prevention of hcv infection |
-
2014
- 2014-01-22 WO PCT/US2014/012610 patent/WO2014116749A1/en active Application Filing
Patent Citations (104)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3773919A (en) | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| USRE30985E (en) | 1978-01-01 | 1982-06-29 | Serum-free cell culture media | |
| US4419446A (en) | 1980-12-31 | 1983-12-06 | The United States Of America As Represented By The Department Of Health And Human Services | Recombinant DNA process utilizing a papilloma virus DNA as a vector |
| EP0073657A1 (en) | 1981-08-31 | 1983-03-09 | Genentech, Inc. | Preparation of hepatitis B surface antigen in yeast |
| US4601978A (en) | 1982-11-24 | 1986-07-22 | The Regents Of The University Of California | Mammalian metallothionein promoter system |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| US4657866A (en) | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US4965199A (en) | 1984-04-20 | 1990-10-23 | Genentech, Inc. | Preparation of functional human factor VIII in mammalian cells using methotrexate based selection |
| US4737456A (en) | 1985-05-09 | 1988-04-12 | Syntex (U.S.A.) Inc. | Reducing interference in ligand-receptor binding assays |
| WO1987000195A1 (en) | 1985-06-28 | 1987-01-15 | Celltech Limited | Animal cell culture |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US6982321B2 (en) | 1986-03-27 | 2006-01-03 | Medical Research Council | Altered antibodies |
| US4927762A (en) | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| US5500362A (en) | 1987-01-08 | 1996-03-19 | Xoma Corporation | Chimeric antibody with specificity to human B cell surface antigen |
| US5648260A (en) | 1987-03-18 | 1997-07-15 | Scotgen Biopharmaceuticals Incorporated | DNA encoding antibodies with altered effector functions |
| US5624821A (en) | 1987-03-18 | 1997-04-29 | Scotgen Biopharmaceuticals Incorporated | Antibodies with altered effector functions |
| WO1990003430A1 (en) | 1988-09-23 | 1990-04-05 | Cetus Corporation | Cell culture medium for enhanced cell growth, culture longevity and product expression |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| WO1990013646A1 (en) | 1989-04-28 | 1990-11-15 | Transgene S.A. | Application of novel dna fragments as a coding sequence for a signal peptide for the secretion of mature proteins by recombinant yeast, expression cassettes, transformed yeasts and corresponding process for the preparation of proteins |
| EP0404097A2 (en) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispecific and oligospecific, mono- and oligovalent receptors, production and applications thereof |
| US6417429B1 (en) | 1989-10-27 | 2002-07-09 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US6075181A (en) | 1990-01-12 | 2000-06-13 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5770429A (en) | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| US5750373A (en) | 1990-12-03 | 1998-05-12 | Genentech, Inc. | Enrichment method for variant proteins having altered binding properties, M13 phagemids, and growth hormone variants |
| US5571894A (en) | 1991-02-05 | 1996-11-05 | Ciba-Geigy Corporation | Recombinant antibodies specific for a growth factor receptor |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| WO1993001161A1 (en) | 1991-07-11 | 1993-01-21 | Pfizer Limited | Process for preparing sertraline intermediates |
| US5648237A (en) | 1991-09-19 | 1997-07-15 | Genentech, Inc. | Expression of functional antibody fragments |
| US5587458A (en) | 1991-10-07 | 1996-12-24 | Aronex Pharmaceuticals, Inc. | Anti-erbB-2 antibodies, combinations thereof, and therapeutic and diagnostic uses thereof |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| WO1993016185A2 (en) | 1992-02-06 | 1993-08-19 | Creative Biomolecules, Inc. | Biosynthetic binding protein for cancer marker |
| WO1994011026A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Therapeutic application of chimeric and radiolabeled antibodies to human b lymphocyte restricted differentiation antigen for treatment of b cell lymphoma |
| WO1994029351A2 (en) | 1993-06-16 | 1994-12-22 | Celltech Limited | Antibodies |
| US5789199A (en) | 1994-11-03 | 1998-08-04 | Genentech, Inc. | Process for bacterial production of polypeptides |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US5840523A (en) | 1995-03-01 | 1998-11-24 | Genetech, Inc. | Methods and compositions for secretion of heterologous polypeptides |
| US5869046A (en) | 1995-04-14 | 1999-02-09 | Genentech, Inc. | Altered polypeptides with increased half-life |
| US6267958B1 (en) | 1995-07-27 | 2001-07-31 | Genentech, Inc. | Protein formulation |
| WO1997030087A1 (en) | 1996-02-16 | 1997-08-21 | Glaxo Group Limited | Preparation of glycosylated antibodies |
| US6171586B1 (en) | 1997-06-13 | 2001-01-09 | Genentech, Inc. | Antibody formulation |
| WO1998058964A1 (en) | 1997-06-24 | 1998-12-30 | Genentech, Inc. | Methods and compositions for galactosylated glycoproteins |
| WO1999022764A1 (en) | 1997-10-31 | 1999-05-14 | Genentech, Inc. | Methods and compositions comprising glycoprotein glycoforms |
| US7189826B2 (en) | 1997-11-24 | 2007-03-13 | Institute For Human Genetics And Biochemistry | Monoclonal human natural antibodies |
| US7087409B2 (en) | 1997-12-05 | 2006-08-08 | The Scripps Research Institute | Humanization of murine antibody |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| WO1999051642A1 (en) | 1998-04-02 | 1999-10-14 | Genentech, Inc. | Antibody variants and fragments thereof |
| US6602684B1 (en) | 1998-04-20 | 2003-08-05 | Glycart Biotechnology Ag | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US6040498A (en) | 1998-08-11 | 2000-03-21 | North Caroline State University | Genetically engineered duckweed |
| US7371826B2 (en) | 1999-01-15 | 2008-05-13 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US7332581B2 (en) | 1999-01-15 | 2008-02-19 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2000061739A1 (en) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| US6420548B1 (en) | 1999-10-04 | 2002-07-16 | Medicago Inc. | Method for regulating transcription of foreign genes |
| US7125978B1 (en) | 1999-10-04 | 2006-10-24 | Medicago Inc. | Promoter for regulating expression of foreign genes |
| WO2001029246A1 (en) | 1999-10-19 | 2001-04-26 | Kyowa Hakko Kogyo Co., Ltd. | Process for producing polypeptide |
| US20070117126A1 (en) | 1999-12-15 | 2007-05-24 | Genentech, Inc. | Shotgun scanning |
| US20060025576A1 (en) | 2000-04-11 | 2006-02-02 | Genentech, Inc. | Multivalent antibodies and uses therefor |
| US20030115614A1 (en) | 2000-10-06 | 2003-06-19 | Yutaka Kanda | Antibody composition-producing cell |
| US20020164328A1 (en) | 2000-10-06 | 2002-11-07 | Toyohide Shinkawa | Process for purifying antibody |
| WO2002031140A1 (en) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cells producing antibody compositions |
| US20070061900A1 (en) | 2000-10-31 | 2007-03-15 | Murphy Andrew J | Methods of modifying eukaryotic cells |
| US7041870B2 (en) | 2000-11-30 | 2006-05-09 | Medarex, Inc. | Transgenic transchromosomal rodents for making human antibodies |
| WO2003011878A2 (en) | 2001-08-03 | 2003-02-13 | Glycart Biotechnology Ag | Antibody glycosylation variants having increased antibody-dependent cellular cytotoxicity |
| US20030157108A1 (en) | 2001-10-25 | 2003-08-21 | Genentech, Inc. | Glycoprotein compositions |
| US20040093621A1 (en) | 2001-12-25 | 2004-05-13 | Kyowa Hakko Kogyo Co., Ltd | Antibody composition which specifically binds to CD20 |
| US20040132140A1 (en) | 2002-04-09 | 2004-07-08 | Kyowa Hakko Kogyo Co., Ltd. | Production process for antibody composition |
| WO2003084570A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | DRUG CONTAINING ANTIBODY COMPOSITION APPROPRIATE FOR PATIENT SUFFERING FROM FcϜRIIIa POLYMORPHISM |
| WO2003085119A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | METHOD OF ENHANCING ACTIVITY OF ANTIBODY COMPOSITION OF BINDING TO FcϜ RECEPTOR IIIa |
| US20040110282A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells in which activity of the protein involved in transportation of GDP-fucose is reduced or lost |
| US20040110704A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells of which genome is modified |
| US20040109865A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Antibody composition-containing medicament |
| WO2003085107A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Cells with modified genome |
| US20050119455A1 (en) | 2002-06-03 | 2005-06-02 | Genentech, Inc. | Synthetic antibody phage libraries |
| US20050014934A1 (en) | 2002-10-15 | 2005-01-20 | Hinton Paul R. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| WO2004056312A2 (en) | 2002-12-16 | 2004-07-08 | Genentech, Inc. | Immunoglobulin variants and uses thereof |
| US20050079574A1 (en) | 2003-01-16 | 2005-04-14 | Genentech, Inc. | Synthetic antibody phage libraries |
| US20060104968A1 (en) | 2003-03-05 | 2006-05-18 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminogly ycanases |
| US20050260186A1 (en) | 2003-03-05 | 2005-11-24 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| WO2005035586A1 (en) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | Fused protein composition |
| WO2005035778A1 (en) | 2003-10-09 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | PROCESS FOR PRODUCING ANTIBODY COMPOSITION BY USING RNA INHIBITING THE FUNCTION OF α1,6-FUCOSYLTRANSFERASE |
| US20050123546A1 (en) | 2003-11-05 | 2005-06-09 | Glycart Biotechnology Ag | Antigen binding molecules with increased Fc receptor binding affinity and effector function |
| WO2005053742A1 (en) | 2003-12-04 | 2005-06-16 | Kyowa Hakko Kogyo Co., Ltd. | Medicine containing antibody composition |
| US7527791B2 (en) | 2004-03-31 | 2009-05-05 | Genentech, Inc. | Humanized anti-TGF-beta antibodies |
| US20050266000A1 (en) | 2004-04-09 | 2005-12-01 | Genentech, Inc. | Variable domain library and uses |
| WO2005100402A1 (en) | 2004-04-13 | 2005-10-27 | F.Hoffmann-La Roche Ag | Anti-p-selectin antibodies |
| WO2006029879A2 (en) | 2004-09-17 | 2006-03-23 | F.Hoffmann-La Roche Ag | Anti-ox40l antibodies |
| US7521541B2 (en) | 2004-09-23 | 2009-04-21 | Genetech Inc. | Cysteine engineered antibodies and conjugates |
| WO2006044908A2 (en) | 2004-10-20 | 2006-04-27 | Genentech, Inc. | Antibody formulation in histidine-acetate buffer |
| WO2006100449A1 (en) | 2005-03-19 | 2006-09-28 | Medical Research Council | Improvements in or relating to treatment and prevention of viral infections |
| US20070160598A1 (en) | 2005-11-07 | 2007-07-12 | Dennis Mark S | Binding polypeptides with diversified and consensus vh/vl hypervariable sequences |
| US20070237764A1 (en) | 2005-12-02 | 2007-10-11 | Genentech, Inc. | Binding polypeptides with restricted diversity sequences |
| US20070292936A1 (en) | 2006-05-09 | 2007-12-20 | Genentech, Inc. | Binding polypeptides with optimized scaffolds |
| US20080069820A1 (en) | 2006-08-30 | 2008-03-20 | Genentech, Inc. | Multispecific antibodies |
| WO2008077546A1 (en) | 2006-12-22 | 2008-07-03 | F. Hoffmann-La Roche Ag | Antibodies against insulin-like growth factor i receptor and uses thereof |
| US20090002360A1 (en) | 2007-05-25 | 2009-01-01 | Innolux Display Corp. | Liquid crystal display device and method for driving same |
| WO2009081285A2 (en) | 2007-12-17 | 2009-07-02 | Medical Research Council Technology | Hepatitis c virus antibodies |
| WO2009089004A1 (en) | 2008-01-07 | 2009-07-16 | Amgen Inc. | Method for making antibody fc-heterodimeric molecules using electrostatic steering effects |
| WO2010080528A1 (en) * | 2008-12-17 | 2010-07-15 | Genentech, Inc. | Hepatitis c virus combination therapy |
| WO2011147863A1 (en) * | 2010-05-25 | 2011-12-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Combination of anti-envelope antibodies and anti-receptor antibodies for the treatment and prevention of hcv infection |
Non-Patent Citations (145)
| Title |
|---|
| "Methods in Enzymology", ACADEMIC PRESS, INC. |
| "Short Protocols in Molecular Biology", 1999, WILEY AND SONS |
| A. DOYLE, J.B. GRIFFITHS, AND D.G. NEWELL,: "Cell and Tissue Culture: Laboratory Procedures", 1993, J. WILEY AND SONS |
| ALMAGRO; FRANSSON, FRONT. BIOSCI., vol. 13, 2008, pages 1619 - 1633 |
| ARMBRUSTER ET AL., I. ANTIMICROB. CHEMOLHER., vol. 54, 2004, pages 915 - 920 |
| BACA ET AL., J. BIOL. CHEM., vol. 272, 1997, pages 10678 - 10684 |
| BARNES ET AL., ANAL. RIOC.HEM., vol. 102, 1980, pages 25 - 5 |
| BOERNER ET AL., J. IMMUNOL., vol. 147, 1991, pages 86 |
| BRENNAN ET AL., SCIENCE, vol. 229, 1985, pages 81 |
| BRODEUR ET AL.: "Monoclonal Antibody Production Techniques and Applications", 1987, MARCEL DEKKER, INC., pages: 51 - 63 |
| BRUGGEMANN, M. ET AL., J. EXP. MED., vol. 166, 1987, pages 1351 - 1361 |
| BUKH J ET AL., SEMIN LIV DIS., vol. 15, no. 1, 1995, pages 41 - 63 |
| C.A. JANEWAY AND P. TRAVERS,: "Immunobiology", 1997 |
| CARTER ET AL., BIOFFECHNOLOGY, vol. 10, 1992, pages 163 - 167 |
| CARTER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 89, 1992, pages 4285 |
| CHARLTON: "Methods in Molecular Biology", vol. 248, 2003, HUMANA PRESS, pages: 245 - 254 |
| CHEN ET AL., J. MOL. BIOL., vol. 293, 1999, pages 865 - 881 |
| CHOTHIA; LESK, J. MOL. BIOL., vol. 196, 1987, pages 901 - 917 |
| CHOWDHURY, METHODS MOL. BIOL., vol. 207, 2008, pages 179 - 196 |
| CHUNG ET AL., NUCLEIC ACIDS RES., vol. 16, 1988, pages 3580 |
| CLACKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| CLARKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| CLYNES ET AL., PROC. NAT'LACAD. SCI. USA, vol. 95, 1998, pages 652 - 656 |
| CRAGG, M.S. ET AL., BLOOD, vol. 101, 2003, pages 1045 - 1052 |
| CRAGG, M.S.; M.J. GLENNIE, BLOOD, vol. 103, 2004, pages 2738 - 2743 |
| CUNNINGHAM; WELLS, SCIENCE, vol. 244, 1989, pages 1081 - 1085 |
| D. CATTY.,: "Antibodies: A Practical Approach", 1988, IRL PRESS |
| D. JOHN WALKER: "Methods in Molecular Biology", HUMANA PRESS |
| D.M. WEIR AND C.C. BLACKWELL,: "Handbook of Experimental Immunology", BLACKWELL SCIENTIFIC PUBLICATIONS |
| DALL'ACQUA ET AL., METHODS, vol. 36, 2005, pages 43 - 60 |
| DIAO ET AL., J VIROL., vol. 86, no. 20, 2012, pages 10935 - 49 |
| DIAO ET AL., VIROL., vol. 86, no. 20, 2012, pages 10935 - 49 |
| DUNCAN; WINTER, NATURE, vol. 322, 1988, pages 738 - 40 |
| E. HARLOW; D. LANE: "Using Antibodies: A Laboratory Manual", 1999, COLD SPRING HARBOR LABORATORY PRESS |
| F.M. AUSUBEL, ET AL.: "Current Protocols in Molecular Biology", 2003 |
| FELLOUSE, PROC. NUTL. ACAD. SCI. USA, vol. 101, no. 34, 2004, pages 12467 - 12472 |
| FLATMAN ET AL., J. CHROMATOGR. B, vol. 848, 2007, pages 79 - 87 |
| FLEER ET AL., BIOÍRECHNOLOGY, vol. 9, 1991, pages 968 - 975 |
| GAZZANO-SANTORO ET AL., J. IMMUNOL. METHODS, vol. 202, 1996, pages 163 |
| GEMGROSS, NAT. BIOTECH, vol. 22, 2004, pages 1409 - 1414 |
| GRAHAM ET AL., J. GEN VIROL., vol. 36, 1977, pages 59 |
| GRIFFITHS ET AL., EMBO J, vol. 12, 1993, pages 725 - 734 |
| GRUBER, I. LMMUNOL., vol. 152, 1994, pages 5368 |
| GUSS ET AL., EMBO J., vol. 5, 1986, pages 15671575 |
| GUYER ET AL., J. IMMUNOL., vol. 117, 1976, pages 587 |
| HAM ET AL., METH. FNZ., vol. 58, 1979, pages 44 |
| HARLOW; LANE: "Antibodies, A Laboratory Manual, and Animal Cell Culture", 1987 |
| HARLOW; LANE: "Antibodies: A Laboratory Manual", 1988, COLD SPRING HARBOR LABORATORY |
| HELLSTROM ET AL., PROC. NAT'L ACAD. SCI. USA, vol. 83, 1986, pages 7059 - 7063 |
| HELLSTROM, I ET AL., PROC. NAT'L ACAD. SCI. USA, vol. 82, 1985, pages 1499 - 1502 |
| HOLLINGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 6444 - 6448 |
| HOOGENBOOM ET AL.: "Methods in Molecular Biology", vol. 178, 2001, HUMAN PRESS, pages: 1 - 37 |
| HOOGENBOOM; WINTER, J. MOL. BIOL., vol. 227, 1992, pages 381 - 388 |
| HOTZEL ET AL., PROTEIN ENG DES SEL, vol. 24, 2011, pages 679 - 89 |
| HOTZEL ET AL., PROTEIN ENG DES SEL., vol. 24, 2011, pages 679 - 89 |
| HUDSON ET AL., NAT. MED., vol. 9, 2003, pages 129 - 134 |
| IDUSOGIE ET AL., J. IMMUNOL., vol. 164, 2000, pages 4178 - 4184 |
| J.E. CELLIS,: "Cell Biology: A Laboratory Notebook", 1998, ACADEMIC PRESS |
| J.E. COLIGAN ET AL.,: "Current Protocols in Immunology", 1991 |
| J.M. MILLER AND M.P. CALOS;: "Gene Transfer Vectors for Mammalian Cells", 1987 |
| J.P. MATHER AND P.E. ROBERTS,: "Introduction to Cell and Tissue Culture", 1998, PLENUM PRESS |
| JONES, GENETICS, vol. 85, 1977, pages 12 |
| KABAT ET AL.: "Sequences of Proteins of Immunological Interest", 1991, NATIONAL INSTITUTES OF HEALTH |
| KABAT ET AL.: "Sequences of Proteins of Immunological Interest, Fifth Edition,", vol. 1-3, 1991, NIH PUBLICATION 91-3242 |
| KAM ET AL., PROC. NATL. ACAD. SCI. USA, vol. 102, 2005, pages 11600 - 11605 |
| KANDA, Y. ET AL., BIOTECHNOL. BIOENG., vol. 94, no. 4, 2006, pages 680 - 688 |
| KAPADIA ET AL., J VIROL., vol. 81, 2007, pages 374 - 383 |
| KASHMIRI ET AL., METHODS, vol. 36, 2005, pages 25 - 34 |
| KIM ET AL., J. IMMUNOL., vol. 24, 1994, pages 249 |
| KINDT ET AL.: "Kuby Immunology, 6th ed.,", 2007, W.H. FREEMAN AND CO., pages: 91 |
| KLIMKA ET AL., BR. J. CANCER, vol. 83, 2000, pages 252 - 260 |
| KOSTELNY ET AL., J. IMMUNOL., vol. 148, no. 5, 1992, pages 1547 - 1553 |
| KOZBOR J., IMMUNOL., vol. 133, 1984, pages 3001 |
| KUNKEL ET AL., PNAS, vol. 82, no. 2, 1985, pages 488 - 492 |
| LEE ET AL., J MOL BIOL., vol. 340, 2004, pages 1073 - 93 |
| LEE ET AL., J. IMMUNOL. METHODS, vol. 284, no. 1-2, 2004, pages 119 - 132 |
| LEE ET AL., J. MOL. BIOL., vol. 340, no. 5, 2004, pages 1073 - 1093 |
| LEE ET AL., MOL BIOL., vol. 340, 2004, pages 1073 - 93 |
| LI ET AL., NAT. BIOTECH, vol. 24, 2006, pages 210 - 215 |
| LI ET AL., PROC. NATL. ACAD. SCI. USA, vol. 103, 2006, pages 3557 - 3562 |
| LINDENBACH ET AL., SCIENCE, vol. 309, 2005, pages 623 - 626 |
| LINDMARK ET AL., J. IMMUNOL. METH, vol. 62, 1983, pages 1 - 13 |
| LONBERG, CURR. OPIN. IMMUNOL., vol. 20, 2008, pages 450 - 459 |
| LONBERG, NAT. BIOTECH, vol. 23, 2005, pages 1117 - 1125 |
| M. ZANETTI AND J. D. CAPRA,: "The Antibodies", 1995, HARWOOD ACADEMIC PUBLISHERS |
| M.J. GAIT,: "Oligonucleotide Synthesis", 1984 |
| M.J. MACPHERSON, B.D. HAMES AND G.R. TAYLOR: "PCR 2: A Practical Approach", 1995 |
| MACCALLUM ET AL., J. MOL. BIOL, vol. 262, 1996, pages 732 - 745 |
| MARKS ET AL., J. MOL. BIOL., vol. 222, 1992, pages 581 - 597 |
| MARKS; BRADBURY: "Methods in Molecular Biology", vol. 248, 2003, HUMAN PRESS, pages: 161 - 175 |
| MATHER ET AL., ANNALS N.Y. ACAD. SCI., vol. 383, 1982, pages 44 - 68 |
| MATHER, BIOL. REPROD, vol. 23, 1980, pages 243 - 251 |
| MCCAFFERTY ET AL., NATURE, vol. 348, pages 552 - 554 |
| MILSTEIN; CUELLO, NATURE, vol. 305, 1983, pages 537 |
| MORRIS: "Methods in Molecular Biology", vol. 66, 1996, HUMANA PRESS, article "Epitope Mapping Protocols" |
| MORRISON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 6851 - 6855 |
| MULLIS ET AL.,: "PCR: The Polymerase Chain Reaction", 1994 |
| NI, XIANDAI MIANYIXUE, vol. 26, no. 4, 2006, pages 265 - 268 |
| OKAZAKI ET AL., MOL. BIOL., vol. 336, 2004, pages 1239 - 1249 |
| OSBOURN ET AL., METHODS, vol. 36, 2005, pages 61 - 68 |
| OSOL, A.: "Remington's Pharmaceutical Sciences 16th edition,", 1980 |
| OWSIANKA ET AL., J. GEN. VIROL., vol. 82, 2001, pages 1877 - 83 |
| P. FINCH, ANTIBODIES, 1997 |
| P. SEILER ET AL: "Additive Effect of Neutralizing Antibody and Antiviral Drug Treatment in Preventing Virus Escape and Persistence", JOURNAL OF VIROLOGY, vol. 74, no. 13, 1 July 2000 (2000-07-01), pages 5896 - 5901, XP055087071, ISSN: 0022-538X, DOI: 10.1128/JVI.74.13.5896-5901.2000 * |
| P. SHEPHERD AND C. DEAN,: "Monoclonal Antibodies: A Practical Approach", 2000, OXFORD UNIVERSITY PRESS |
| PADLAN, MOL. LMMUNOL, vol. 28, 1991, pages 489 - 498 |
| PANTUA H ET AL: "The Role of Glycosylation in HCV Resistance to Broadly Neutralizing Antibodies", ABSTRACTS OF THE INTERSCIENCE CONFERENCE ON ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, vol. 52, 2012, & 52ND INTERSCIENCE CONFERENCE ON ANTIMICROBIAL AGENTS AND CHEMOTHERAPY (ICAAC); SAN FRANCISCO, CA, USA; SEPTEMBER 09 -12, 2012, pages V400f, XP009177326, ISSN: 0733-6373 * |
| PANTUA HOMER ET AL: "Glycan Shifting on Hepatitis C Virus (HCV) E2 Glycoprotein Is a Mechanism for Escape from Broadly Neutralizing Antibodies", JOURNAL OF MOLECULAR BIOLOGY, vol. 425, no. 11, June 2013 (2013-06-01), pages 1899 - 1914, XP028539104 * |
| PETKOVA, S.B. ET AL., INT'L. IMMUNOL., vol. 18, no. 12, 2006, pages 1759 - 1769 |
| PIETSCHMANN ET AL., PNAS, vol. 103, 2006, pages 7408 - 7413 |
| PORTOLANO ET AL., J. IMMUNOL., vol. 150, 1993, pages 880 - 887 |
| PRESTA ET AL., CANCER RES., vol. 57, 1997, pages 4593 - 4599 |
| PRESTA ET AL., J. IMMUNOL., vol. 151, 1993, pages 2623 |
| QUEEN ET AL., PROC. NAT'L ACAD. SCI. USA, vol. 86, 1989, pages 10029 - 10033 |
| R.I. FRESHNEY: "Animal Cell Culture", 1987 |
| RAVETCH; KINET, ANNU. REV. IMMUNOL., vol. 9, 1991, pages 457 - 492 |
| REESINIK ET AL., GASTROENTEROLOGY, vol. 131, 2006, pages 997 - 1002 |
| REYES ET AL., NATURE, vol. 297, 1982, pages 598 - 601 |
| RIECHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 329 |
| RIPKA ET AL., ARCH. BIOCHEM. BIOPHYS., vol. 249, 1986, pages 533 - 545 |
| ROSENBURG AND MOORE: "The Pharmacology of Monoclonal Antibodies", vol. 113, 1994, SPRINGER-VERLAG, pages: 269 - 315 |
| ROSOK ET AL., J. BIOL. CHEM., vol. 271, 1996, pages 22611 - 22618 |
| SAMBROOK ET AL.: "Molecular Cloning: A Laboratory Manual 3d edition", 2001, COLD SPRING HARBOR LABORATORY PRESS |
| SHIELDS ET AL., J. BIOL. CHEM., vol. 9, no. 2, 2001, pages 6591 - 6604 |
| SIDHU ET AL., J. MOL. BIOL., vol. 338, no. 2, 2004, pages 299 - 310 |
| SIDHU ET AL.: "Phage Display: A Practical Approach", 2004, OXFORD UNIVERSITY PRESS, pages: 27 - 41 |
| SIMMONDS P., J GEN VIROL., vol. 85, 2004, pages 3173 - 88 |
| SIMS ET AL., J. LMMUNOL., vol. 151, 1993, pages 2296 |
| STIEGLER; KATINGER, J. ANLIMICROB. CHEMOLHER., vol. 51, 2003, pages 757 - 759 |
| STINCHCOMB ET AL., NATURE, vol. 282, 1979, pages 39 |
| TRAUNECKER ET AL., EMBO J., vol. 10, 1991, pages 3655 |
| TUTT ET AL., J. IMMUNOL., vol. 147, 1991, pages 60 |
| URLAUB ET AL., PROC. NATL. ACAD. SCI. USA, vol. 77, 1980, pages 4216 |
| V.T. DEVITA ET AL.,: "Cancer: Principles and Practice of Oncology", 1993, J.B. LIPPINCOTT COMPANY |
| VAN DEN BERG, BIOLFECHNOLOGY, vol. 8, 1990, pages 135 |
| VAN DIJK; VAN DE WINKEL, CURR. OPIN. PHARMACAL., vol. 5, 2001, pages 368 - 74 |
| VOLLMERS; BRANDLEIN, HISTOLOGY AND HISTOPATHOLOGY, vol. 20, no. 3, 2005, pages 927 - 937 |
| VOLLMERS; BRANDLEIN, METHODS AND FINDINGS IN EXPERIMENTAL AND CLINICAL PHARMACOLOGY, vol. 27, no. 3, 2005, pages 185 - 91 |
| WINTER ET AL., ANN. REV. IMMUNOL., vol. 12, 1994, pages 433 - 455 |
| WRIGHT ET AL., TIBTECH, vol. 15, 1997, pages 26 - 32 |
| YAMANE-OHNUKI ET AL., BIOTECH. BIOENG., vol. 87, 2004, pages 614 |
| YAMANE-OHNUKI, BIOTECH. BIOENG., vol. 87, 2004, pages 614 |
| YANIV, NATURE, vol. 297, 1982, pages 17 - 18 |
| YAZAKI; WU: "Methods in Molecular Biology", vol. 248, 2003, HUMANA PRESS, pages: 255 - 268 |
| ZHONG ET AL., PNAS, vol. 102, 2005, pages 9294 - 9 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022153898A1 (en) * | 2021-01-13 | 2022-07-21 | デンカ株式会社 | Method for measuring target antigen, and insoluble particles and target antigen measurement kit used therein |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10066022B2 (en) | Anti-Ly6E antibodies and methods of use | |
| TW201620932A (en) | Anti-influenza B virus hemagglutinin antibodies and methods of use | |
| EP2804629A1 (en) | Anti-lrp5 antibodies and methods of use | |
| WO2022162587A1 (en) | Anti-sars-cov-2 antibodies and use thereof in the treatment of sars-cov-2 infection | |
| MX2013001305A (en) | Anti-mhc antibody anti-viral cytokine fusion protein. | |
| TW202003557A (en) | Anti-dengue virus antibodies having cross-reactivity to zika virus and methods of use | |
| WO2013059531A1 (en) | Anti-gcgr antibodies and uses thereof | |
| JP2019031552A (en) | Methods for treating cancer using pd-1 axis binding antagonists and anti-gpc3 antibodies | |
| WO2014099908A1 (en) | Methods for inhibiting viral infection in transplant patients | |
| US20220242938A1 (en) | Anti-Polyubiquitin Multispecific Antibodies | |
| KR20160111948A (en) | H7n9 influenza a therapies with anti-h7 virus antibodies | |
| US10246519B2 (en) | NSP4 inhibitors and methods of use | |
| EP3302563A1 (en) | Humanized anti-ebola virus glycoprotein antibodies and methods of use | |
| WO2014116749A1 (en) | Anti-hcv antibodies and methods of using thereof | |
| JP2018500882A (en) | Animal model of nephropathy and drug for treating it | |
| US20230340081A1 (en) | Anti-hbv antibodies and methods of use | |
| WO2022263638A1 (en) | Anti-sars-cov-2 antibodies and use thereof in the treatment of sars-cov-2 infection | |
| WO2022115466A1 (en) | Broadly neutralizing antibodies to tick-borne encephalitis and related viruses |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14704023 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14704023 Country of ref document: EP Kind code of ref document: A1 |